Clear Sky Science · en
Covalent inhibitor design confers activity against both GDP- and GTP-bound forms of KRAS G12C
Why this matters for future cancer treatments
Cancers driven by mutations in a gene called KRAS have long been considered some of the toughest to treat. Recently, new drugs that shut down one common mutant form, KRAS G12C, have reached patients and begun to change that picture. This paper explores an even more ambitious idea: can we design drugs that block KRAS G12C in both of its main working modes inside the cell, and would that make cancer treatment faster, stronger, or more durable?
Turning a molecular light switch off in two positions
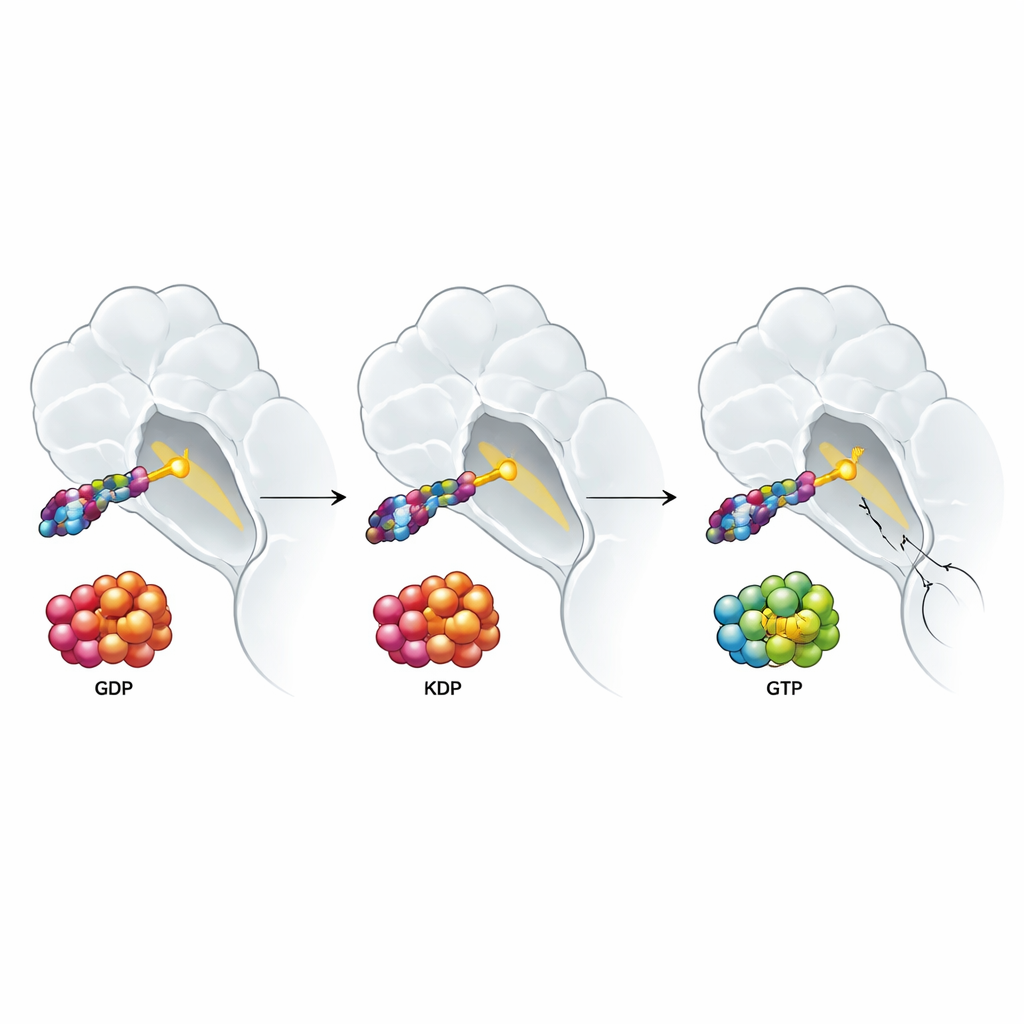
KRAS acts like a tiny on–off switch for cell growth signals. It flips between an “off” state, where it holds one type of energy-carrying molecule (GDP), and an “on” state, where it holds another (GTP). The KRAS G12C mutation sticks this switch partly on and fuels tumor growth. Existing medicines approved by the FDA latch onto the “off” form and lock it in place; over time, as more KRAS cycles through that form, the total pool of mutant protein becomes disabled. Many scientists have argued that a drug able to grab KRAS in both its on and off forms should work better, especially in tumors that adapt by keeping more KRAS in the on state.
Designing a new kind of molecular hook
The authors set out to build such dual-purpose drugs. They focused on a small groove near a flexible region of KRAS called Switch II, where current drugs already bind. Instead of changing how tightly their molecules nestled into that groove, they re-engineered the “warhead” portion that forms a permanent chemical bond to the mutant cysteine at position 12. Through computer modeling and synthesis of dozens of variants, they discovered a special three-part warhead that approaches the cysteine from a slightly different angle. This subtle shift creates enough room for the bulkier GTP molecule, allowing the new compounds to bond to KRAS whether it carries GDP or GTP. Biochemical tests confirmed that lead compounds could disrupt KRAS’s partnership with a key signaling partner, RAF, in both states.
Seeing how the protein reshapes itself
To understand why these molecules worked, the team determined high‑resolution crystal structures of the drug bound to KRAS. These snapshots showed that the new warhead reacts at an unusual site on the chemical scaffold, breaking off a fluorine atom and forming a covalent bond at a different carbon than typical cancer drugs that target cysteines. This altered chemistry helps the drug fit into the Switch II pocket even when GTP is present. The structures also revealed that when the inhibitor binds, it nudges a tiny water molecule and subtly rearranges another loop, Switch I, which normally contacts downstream signaling proteins. This “allosteric” reshaping pulls two amino acids together to form a tight salt bridge, distorting the surface so RAF can no longer dock and pass on growth signals.
Fast shutdown, but no stronger long-term effect
Armed with these structural insights, the researchers refined two lead molecules that efficiently and selectively latch onto KRAS G12C, while largely sparing other proteins with cysteines. In cancer cell lines, these dual‑state inhibitors rapidly blocked a key signaling relay known as the MAPK pathway, as measured by loss of a chemical tag on the ERK protein and strong suppression of cell growth. They inactivated KRAS in cells more quickly than a benchmark drug that only targets the inactive state. In mouse tumor models, one lead compound showed good oral availability, quickly formed covalent bonds to the target, and shrank tumors or slowed their growth. Yet, when the team compared dual‑state and inactive‑state drugs over longer times, both classes eventually achieved similar levels of KRAS shutdown, pathway inhibition, and tumor control.
Why growth signals from the tumor surroundings still win
The study also probed how signals from the tumor’s environment affect these drugs. Growth factors such as EGF and HGF, which are abundant around many tumors, push RAS proteins toward the active GTP‑loaded form and are known to blunt responses to targeted therapies. One might expect a drug that binds the active form of KRAS G12C to sidestep this problem. Instead, the authors found that both dual‑state and inactive‑state inhibitors lost potency when such growth factors were present. Detailed experiments pointed to a surprising culprit: activation of the normal, non‑mutated forms of RAS (H‑RAS and N‑RAS), which can bypass KRAS G12C and keep growth signals flowing. When the researchers deleted H‑RAS and N‑RAS in a lung cancer cell line, growth factor–driven resistance largely disappeared for all types of KRAS G12C‑targeting drugs, whereas a drug that blocks a downstream step in the pathway was less affected in the first place.
What this means for patients and drug development
Overall, the work demonstrates that it is chemically and structurally feasible to build covalent KRAS G12C drugs that grab the protein in both its on and off states, and that these dual‑state inhibitors can shut down signaling more quickly than existing medicines. However, the rapid engagement did not translate into clearly better tumor control or a solution to growth factor–driven resistance in preclinical models. For patients, this suggests that simply adding activity against the active KRAS form may not be enough; combination strategies that also address other RAS family members or downstream signaling nodes may be required. The new warhead and structural blueprint presented here nonetheless expand the toolkit for targeting KRAS and will inform future generations of precision cancer therapies.
Citation: Condakes, M.L., Zhang, Z., Danahy, D.B. et al. Covalent inhibitor design confers activity against both GDP- and GTP-bound forms of KRAS G12C. Nat Commun 17, 2233 (2026). https://doi.org/10.1038/s41467-026-69003-0
Keywords: KRAS G12C, covalent inhibitor, dual-state inhibition, MAPK signaling, drug resistance