Clear Sky Science · en
OmiGA for ultra-efficient molecular quantitative trait loci mapping
Why this matters for health and breeding
Modern genetics has revealed millions of DNA differences that subtly shape traits like disease risk, growth, and metabolism. Most of these differences act not by changing proteins directly, but by fine‑tuning the activity of genes. To understand this regulatory layer, scientists map “molecular traits” such as gene expression back to the genome. This paper introduces OmiGA, a new analysis toolkit that makes this kind of mapping both more accurate and dramatically faster, especially in populations where many individuals are related, such as farm animals and some human families. 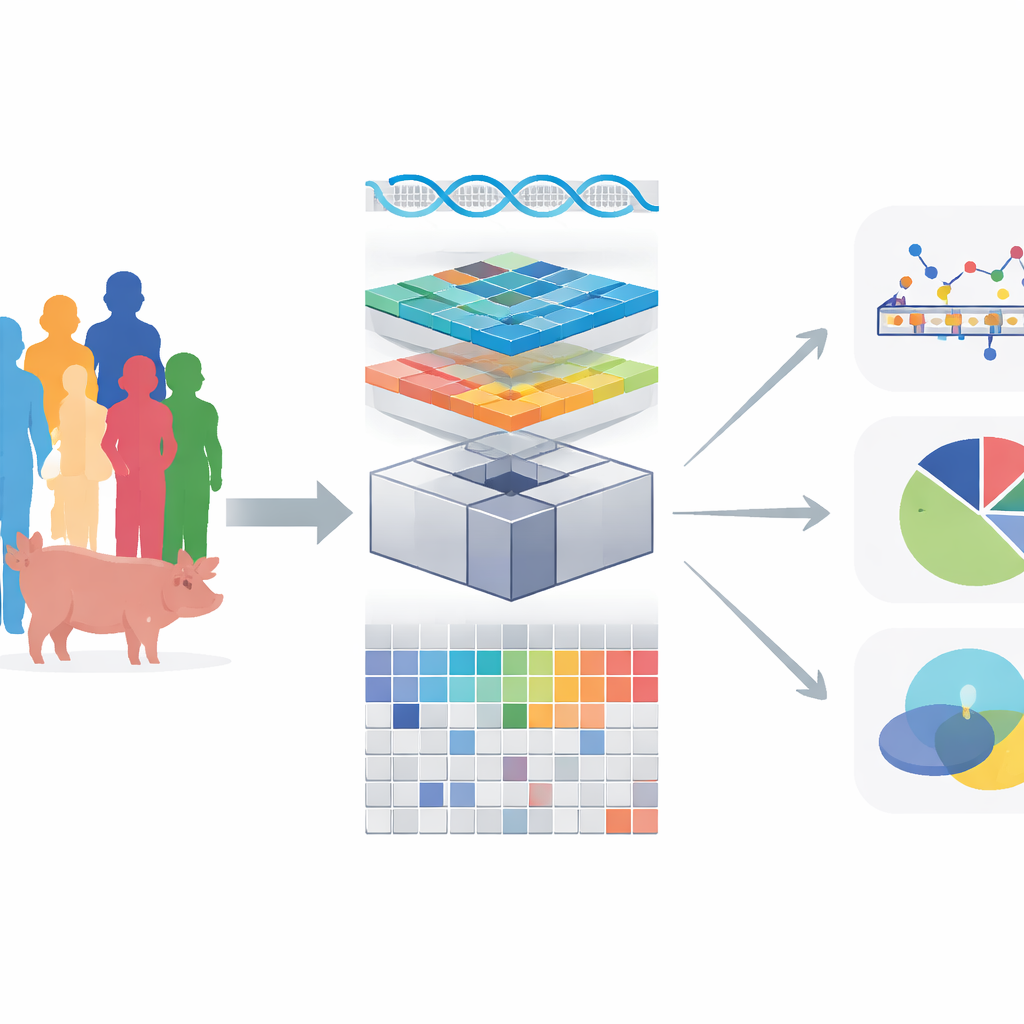
From DNA to switches that control genes
Instead of looking only at outward traits like height or fat content, molecular trait mapping asks how DNA variants change internal readouts: which genes are turned up or down, how RNA is spliced, and similar measurements across thousands of genes and tissues. Sites in the genome that influence these molecular measurements are called molecular quantitative trait loci, or molQTLs. Finding them helps scientists trace a path from DNA change to gene regulation to disease or productivity traits. However, commonly used tools simplify the statistics to keep calculations manageable. They often ignore how closely related individuals are, or how whole stretches of the genome are inherited together, which can produce false signals and hide real effects.
Why relatedness is a statistical headache
In many animal breeds and in human family studies, individuals share large segments of DNA because of recent common ancestors. This “complex relatedness” can make two distant genetic sites seem connected to the same molecular trait simply because they are inherited together, not because both truly regulate the gene. Standard linear models try to patch over this by adding a few summary measures of ancestry, but they struggle when long‑range correlations in the genome are strong. The more related the population and the denser the genetic data, the more these shortcuts inflate apparent signal strength, driving up the rate of false discoveries.
A tailored engine for omics‑scale genetics
OmiGA is built around linear mixed models, a class of statistical tools designed to handle relatedness by explicitly modeling background genetic similarity among individuals. The authors re‑engineered these models for “omics” data, where tens of thousands of molecular traits are tested against millions of DNA variants. They introduce new algorithms that avoid the slowest steps of standard methods, reuse heavy calculations across many traits, and can run on graphics processors for extra speed. OmiGA also estimates how much of each molecular trait is explained by nearby DNA changes, distant regions, and by non‑additive effects where gene copies interact in more complex ways. Together, these features turn a previously cumbersome approach into a practical workhorse for large studies. 
Sharper signals in simulations and real datasets
The team compared OmiGA to popular tools such as tensorQTL, APEX, GCTA, and LDAK using both simulated data and real measurements from pigs and humans. In simulations mimicking closely related pig populations and more loosely related human cohorts, OmiGA consistently kept background noise under control while maintaining or increasing the rate of true discoveries. In real pig tissue data, OmiGA identified substantially more genes whose expression is clearly linked to nearby DNA variants, and did so with lower computational cost. It also produced narrower sets of likely causal variants when zooming in on specific regions, and showed stronger agreement between molecular signals and traditional trait association results, suggesting it is better at pinpointing the true regulatory changes behind complex traits.
New views of dominance and context effects
Beyond standard “additive” effects where each gene copy contributes independently, OmiGA can model dominant effects, where one copy can mask or enhance the other. Applying this to human cell data, the authors found that many genes with classical effects also harbor hidden dominant influences, and in some cases dominant regulation appears where additive effects do not. OmiGA also detects context‑dependent regulation, such as genetic effects that differ with ancestry or environment, and partitions heritability into local and distant components. These capabilities open the door to a richer picture of how DNA variation shapes molecular biology in diverse populations.
What this means going forward
For non‑specialists, the key message is that OmiGA offers a more reliable microscope for seeing how DNA differences tune gene activity, especially in real‑world populations where relatives are common. By reducing false signals and highlighting truly causal variants, it helps link molecular changes to traits like disease risk or meat quality more confidently. This, in turn, can sharpen follow‑up experiments, improve breeding decisions in agriculture, and strengthen efforts to interpret human genetic studies by revealing exactly which regulatory switches in the genome matter most.
Citation: Teng, J., Zhang, W., Gong, W. et al. OmiGA for ultra-efficient molecular quantitative trait loci mapping. Nat Commun 17, 2680 (2026). https://doi.org/10.1038/s41467-026-68978-0
Keywords: molecular QTL mapping, gene expression regulation, linear mixed models, genetic relatedness, omics toolkit