Clear Sky Science · en
FANCD2 restrains fork progression and prevents fragility at early origins upon re-replication
When DNA Copies Go Slightly Off Script
Every time a cell divides, it must copy its entire DNA library exactly once. If parts of that library are copied twice, or copied in a rushed and sloppy way, the result can be broken chromosomes and mutations that fuel cancer. This study looks at what happens when the cell’s safeguards against extra rounds of copying start to fail, and reveals how a repair protein called FANCD2 steps in to keep mildly misbehaving cells from sliding into full-blown genomic chaos.
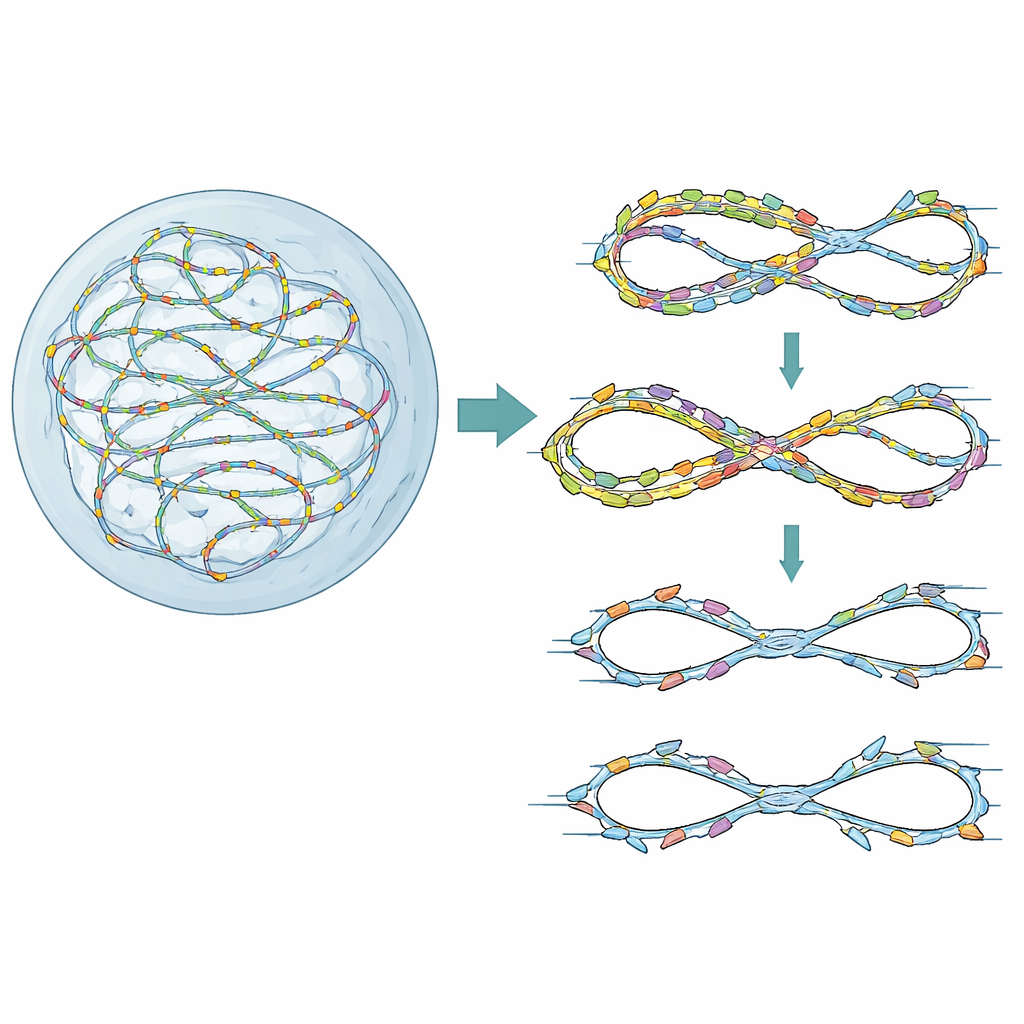
Guardrails for One Clean Copy
Our chromosomes are duplicated from thousands of starting points, or “origins,” that are licensed and then fired in a carefully timed sequence. A small protein named Geminin normally helps ensure that each origin fires only once per cell cycle. When Geminin is lost or weakened, some origins can fire again on already copied DNA, a situation known as re-replication. Cancer cells, which often overproduce licensing factors, are especially prone to this problem. The authors first used a high-content genetic screen in human cells primed for low-level re-replication by Geminin depletion. They asked which DNA repair and checkpoint genes become crucial in this stressed state and found that FANCD2, best known for repairing DNA crosslinks in Fanconi anemia, emerged as a key protector of cell survival and genome integrity.
A First Responder at Overworked Copying Machines
The team then tracked where and when FANCD2 appears in cells undergoing re-replication. Shortly after Geminin is removed, FANCD2 rapidly accumulates on chromatin and forms bright nuclear foci, well before widespread DNA breakage is detectable. Using labeling of newly synthesized DNA together with proximity assays, they showed that FANCD2 is recruited directly to active replication machines, especially in cells whose DNA is already being copied a second time. In synchronized cells released into the next division cycle, a distinct population showing a diffuse, over-replication DNA pattern emerged. These cells displayed strong FANCD2 and RPA signals, indicating ongoing replication stress, and were held at the boundary before mitosis by an active checkpoint, suggesting that FANCD2 is part of an early response that stabilizes stressed forks rather than simply reacting to broken DNA.
Holding Back Runaway Forks and Hidden Gaps
To test how FANCD2 shapes DNA copying, the researchers combined Geminin loss with FANCD2 depletion. Surprisingly, removing FANCD2 did not increase the fraction of cells with obviously re-replicated genomes. Instead, single-molecule DNA fiber assays revealed that replication forks traveled farther and became more asymmetric, a sign of uneven and unstable progression. These faster forks left behind more single-stranded gaps in the newly made DNA, seen as intense RPA and native BrdU foci and confirmed by the sensitivity of labeled tracts to an enzyme that cuts single-stranded regions. Cells lacking both Geminin and FANCD2 showed a jump in chromosome breaks, fragments, nuclear bodies, and micronuclei, all hallmarks of severe genomic instability. Blocking PARP, a factor that normally helps manage such gaps, mimicked and worsened these defects, underscoring that uncontrolled gap formation is central to the damage.
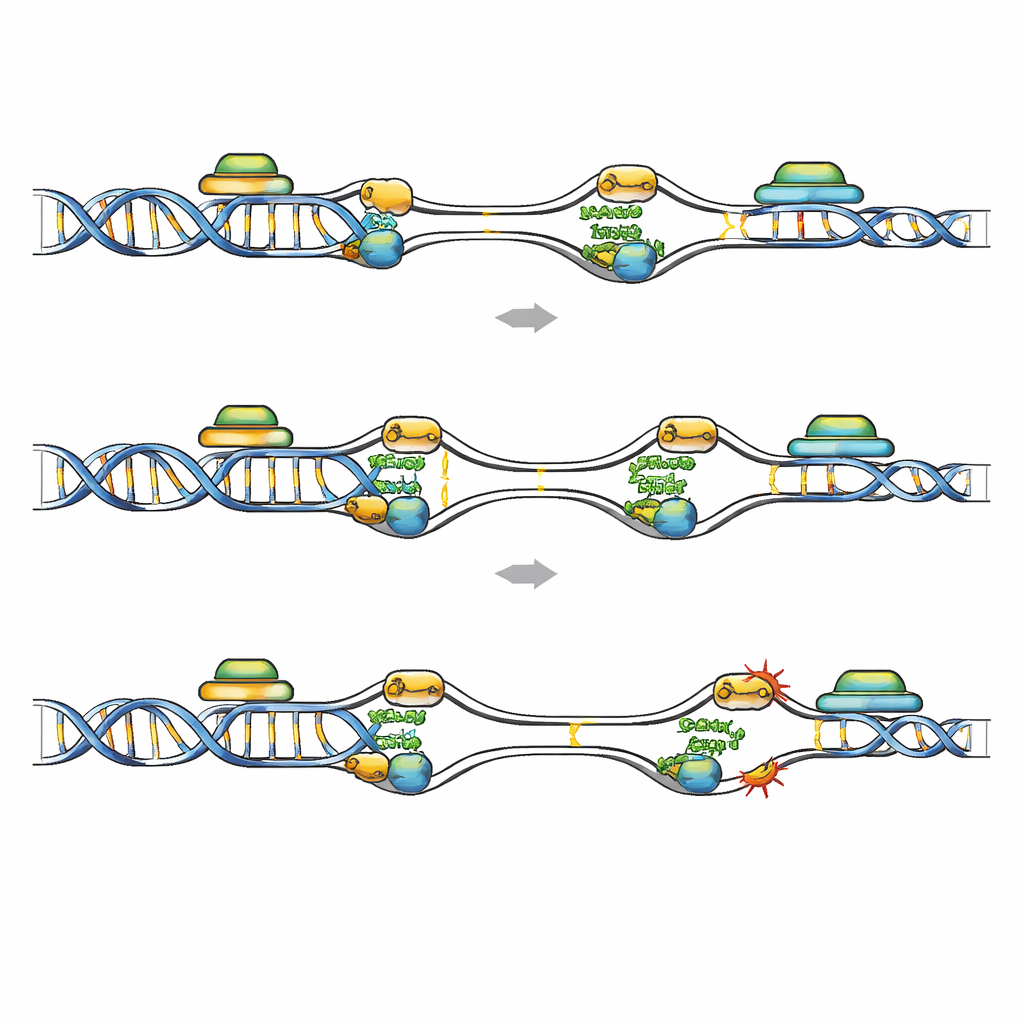
Fragile Hotspots Where Copying and Reading Collide
Genome-wide mapping of FANCD2 binding offered a view of where re-replication is most dangerous. In leukemic cells depleted of Geminin, FANCD2 shifted from classical common fragile sites to early-firing replication origins embedded within short, GC-rich, highly transcribed genes. These regions carry marks of active transcription and are prone to R-loops, where nascent RNA hybrids with its DNA template, potentially blocking replication. Public datasets showed more DNA damage and elevated RNA–DNA hybrid signals in FANCD2-enriched genes after Geminin loss, and these regions overlapped with so-called early replicating fragile sites. When transcription was broadly dampened with a drug, or when R-loops were specifically removed by overexpressing RNase H1, the number of FANCD2, RPA, and DNA damage foci in Geminin-deficient cells dropped markedly. This indicates that collisions between re-fired origins and active transcription units, amplified by R-loops, create fragile hotspots that FANCD2 must protect.
Fine-Tuning Protection Through Chemical Tags
FANCD2 is activated in part by the attachment of a small ubiquitin-like tag. By depleting FANCA, a core component of the tagging machinery, and by using cells that express a FANCD2 mutant resistant to this modification, the authors showed that mono-ubiquitination improves survival of re-replicating cells but is not absolutely required. Even non-tagged FANCD2 provided partial protection, consistent with distinct roles in both sensing and stabilizing stressed forks. The overall picture is that FANCD2 helps slow and organize replication at vulnerable early origins and limits how many and how large the single-stranded gaps become.
Why This Matters for Cancer Treatment
For non-specialists, the core message is that not all replication mistakes are catastrophic from the start. Mild re-replication, as occurs in some tumors, can be tolerated if protective systems like FANCD2 keep runaway DNA copying in check and prevent fragile gaps from turning into broken chromosomes. When this safeguard is removed or overwhelmed, the same low-level licensing errors rapidly escalate into genome shattering. Because Geminin loss and replication licensing defects are enriched in cancer cells, and many tumors already carry weaknesses in the Fanconi/BRCA network, the vulnerabilities uncovered here suggest therapeutic strategies: combining inhibitors that push cancer cells toward re-replication with drugs that exacerbate gap accumulation, such as PARP inhibitors, could selectively drive malignant cells past their tolerance limit while sparing normal cells with intact protection.
Citation: Badra-Fajardo, N., Karydi, E., Bayona-Feliu, A. et al. FANCD2 restrains fork progression and prevents fragility at early origins upon re-replication. Nat Commun 17, 2478 (2026). https://doi.org/10.1038/s41467-026-68966-4
Keywords: DNA replication stress, FANCD2, Geminin, re-replication, genome instability