Clear Sky Science · en
Developments and challenges in hit progression within fragment-based drug discovery
Turning Tiny Chemical Pieces into Future Medicines
Modern medicines are often discovered by sifting through huge collections of molecules, a process that is slow, expensive and increasingly inefficient. This article explores a newer approach called fragment-based drug discovery, which starts from very small chemical pieces and builds them up step by step into promising drug candidates. For readers, it offers a window into how smarter design, automation and artificial intelligence could make tomorrow’s treatments faster to find and more widely available.
Why Start Small Instead of Screening Everything
Traditional drug discovery often relies on testing millions of fairly large molecules to see which ones stick to a disease-related protein. Fragment-based methods take the opposite route: they screen a much smaller collection of tiny molecules, or “fragments,” each representing a simple chemical shape. These fragments bind only weakly, but because they are so small and diverse, they explore chemical possibilities far more efficiently. The challenge is that weak signals are hard to detect and interpret, so researchers need very sensitive experiments and careful cross-checks to be sure that a fragment really binds and is not just an assay artefact. Structural techniques like X-ray crystallography and cryo-electron microscopy can reveal exactly how a fragment sits in a protein pocket, while solution methods such as NMR, calorimetry and surface plasmon resonance measure how strongly and how fast it binds.
From First Hits to Promising Leads
Once useful fragments are found, the real work begins: turning these faint “hits” into strong, selective “lead” compounds. The article frames this journey as repeated “Design, Make, Test” cycles. In the Design step, chemists and computers propose ways to grow, link or merge fragments so they fill the protein pocket better, avoid unwanted reactivity and maintain good physical properties like solubility. In the Make step, these designs are synthesized, increasingly with the help of robots, high-throughput chemistry and clever route-planning software. The Test step then measures whether the new molecules truly bind better, work on the intended biological function and avoid common pitfalls such as pan-assay interference compounds that give misleading signals. Because fragments start weak, several rounds of this loop are often needed before compounds become strong enough to resemble real drug candidates.
New Tools: Automation, AI and Smart Libraries
The review highlights how a new generation of tools is reshaping each stage of this cycle. Fragment libraries are now designed not only to be diverse, but also to be “synthetically sociable,” meaning they can be easily expanded in many directions using robust reactions. Specialized sets of fragments target particular protein families, metal-containing sites or even form covalent bonds with specific amino acids, helping to tackle previously “undruggable” targets. On the digital side, artificial intelligence models and physics-based simulations help suggest which chemical changes might improve binding or reduce toxicity, and can sift through ultra-large virtual spaces of billions of possible molecules. These predictions are increasingly combined with active learning loops, where a small number of expensive simulations or experiments trains faster models that can guide the next wave of designs.
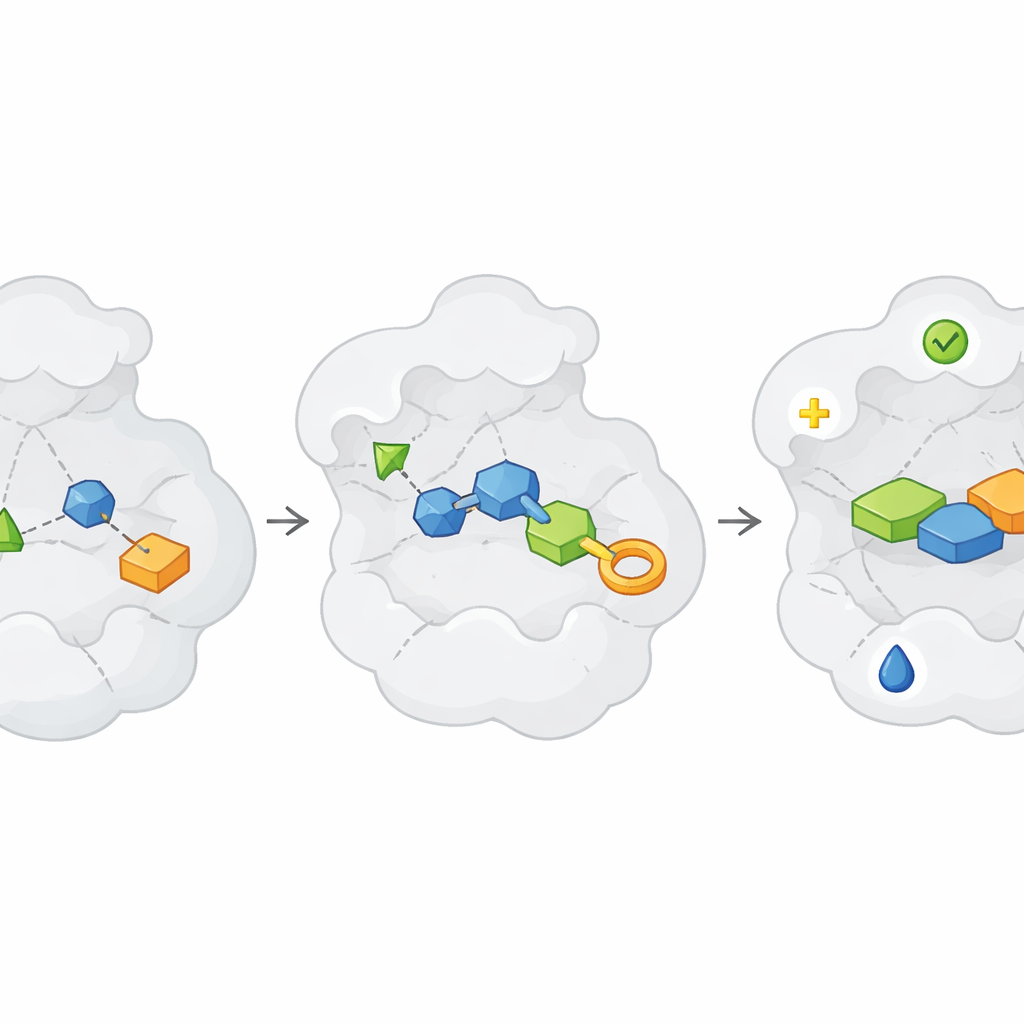
Making and Testing at Scale Without Slowing Down
A major bottleneck in drug discovery is simply making and purifying enough compounds to test. The article describes how high-throughput synthesis robots, flow chemistry and new extraction methods can churn out hundreds or thousands of related molecules around a fragment hit. Some approaches even skip full purification at first: crude reaction mixtures are tested directly in sensitive assays such as crystallography, kinetic measurements or NMR, an approach sometimes called “direct-to-biology.” Quality checks like mass spectrometry are used in parallel to keep track of which mixtures actually contain the intended product. Although the data can be noisy, combining these rapid tests with smart analytics and follow-up clean synthesis allows researchers to map out structure–activity relationships much faster than with traditional one-by-one chemistry.
What This Means for Future Medicines
Overall, the article concludes that fragment-based drug discovery has matured into a powerful and flexible strategy for finding new medicines, especially when paired with modern automation and AI. Starting from tiny, efficient building blocks lets scientists explore chemical space more thoughtfully, but demands careful validation because the starting signals are so weak. The authors argue that the greatest gains will come from tightly integrating design, synthesis and testing into semi-automated, data-driven workflows, while sharing fragment data and methods openly so that even resource-limited groups can benefit. If these developments continue, fragment-based approaches could help reverse the long-term decline in research productivity and speed the arrival of safer, more effective drugs for a wide range of diseases.
Citation: Grosjean, H., Biggin, P.C. Developments and challenges in hit progression within fragment-based drug discovery. Nat Commun 17, 2226 (2026). https://doi.org/10.1038/s41467-026-68941-z
Keywords: fragment-based drug discovery, hit-to-lead optimization, design-make-test cycle, high-throughput screening, computational drug design