Clear Sky Science · en
A neurotoxic cryptic peptide arising from TDP-43-dependent cryptic splicing of PKN1
Hidden threats inside brain cells
Many brain diseases, including amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease, involve clumps of a protein called TDP-43. Scientists know that when this protein stops working properly, nerve cells lose vital messages and eventually die. This study reveals a more surprising twist: TDP-43 failure can also cause brain cells to manufacture a previously unknown, toxic mini‑protein, which in turn damages memory circuits. Understanding this hidden player may open new routes for diagnosis and treatment in devastating dementias.
How a cellular proofreader keeps RNA in line
Inside neurons, TDP-43 acts like a proofreader for RNA, the intermediate messages that sit between DNA and proteins. It binds to specific short sequences and blocks the insertion of stray “extra pieces” into these messages. When TDP-43 is lost or mislocalized, as in ALS and many Alzheimer’s cases, these extra pieces—called cryptic exons—can slip into RNA. Until now, most known cryptic exons simply caused a loss of normal protein by making the message unstable and quickly destroyed. Whether such events could also generate new, harmful proteins was not clear.
A cryptic splice creates a toxic fragment
The authors focused on a gene called PKN1, which helps maintain the internal scaffolding and signal flow of neurons. Using cell models where TDP-43 was reduced, they discovered a previously unrecognized cryptic exon, dubbed PKN1‑5a1, inserted between two normal segments of the PKN1 RNA. This insertion introduces an early stop signal, producing a shortened RNA. Remarkably, part of this faulty message escapes the cell’s quality‑control system and is translated into a stable fragment of the PKN1 protein containing only its first 207 amino acids. The team named this truncated product PKN207. They showed that TDP-43 normally prevents this mistake by binding to several UG‑rich regions flanking the cryptic exon; when that binding is lost, the exon is spliced in and PKN207 is produced.
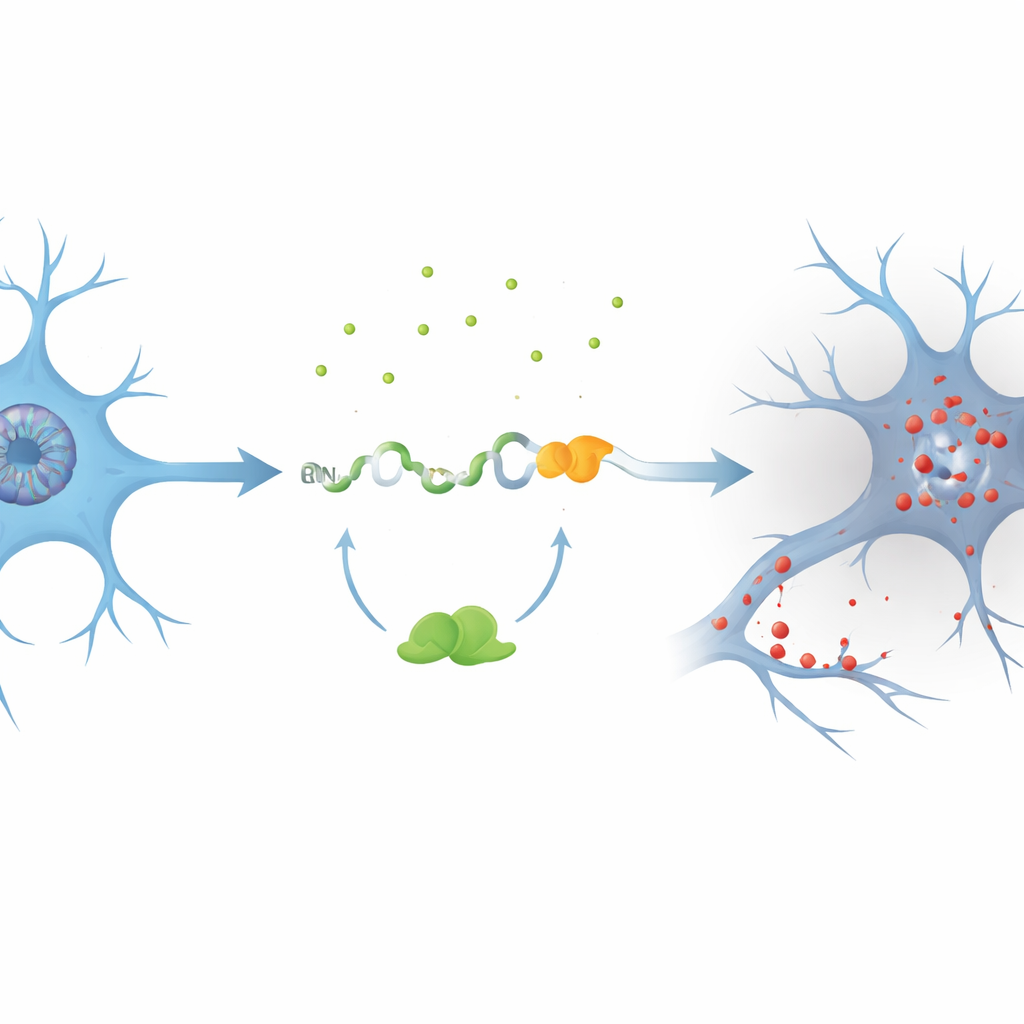
Evidence from patient brains and large datasets
To see whether this event happens in human disease, the researchers mined RNA sequencing data from hundreds of ALS brain and spinal cord samples. They found widespread activation of the PKN1‑5a1 cryptic exon in regions known to be affected by TDP-43 pathology, such as motor cortex and spinal cord, but not in the relatively spared cerebellum. They then generated highly specific antibodies that recognize only the unique tail of PKN207, not the full-length PKN1 protein. In hippocampal tissue from Alzheimer’s patients who also showed abnormal, phosphorylated TDP-43, these antibodies detected a distinct band corresponding to PKN207, while such a band was absent in control brains. Additional Alzheimer’s datasets confirmed that the cryptic exon is turned on even at early disease stages, suggesting that this molecular error may start long before symptoms become obvious.
A mini‑protein with major impact on memory
Finding PKN207 in diseased human brain raised the key question: is it harmful? To test this, the team used viruses to drive production of either normal PKN1 or PKN207 specifically in the hippocampus—a brain area critical for memory—of young mice. Months later, both groups of mice showed impaired learning in the Morris water maze, swimming longer to find a hidden platform. Their spinal fluid contained higher levels of a structural protein, neurofilament light chain, a marker of axonal damage. In cultured neurons, boosting PKN207 triggered cell injury, as measured by leakage of an enzyme that signals membrane damage. Detailed protein profiling of the hippocampus revealed broad changes in pathways linked to synaptic strength (long‑term potentiation) and well‑known neurodegenerative diseases, with especially strong disruption of molecules that support efficient signaling and a healthy nerve‑fiber scaffold.
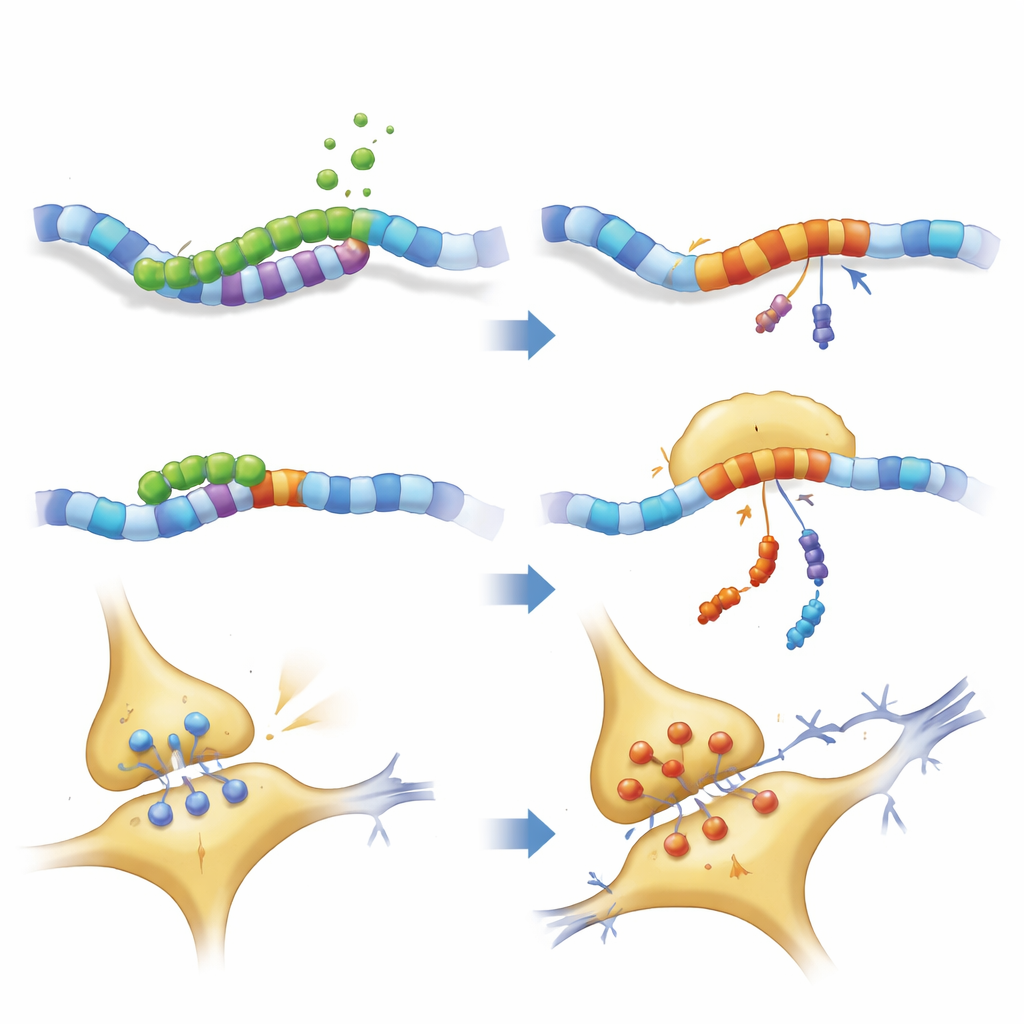
How the fragment disrupts brain wiring
Closer examination of neuronal structure showed that both full-length PKN1 and PKN207 disturbed the neurofilament network that gives axons their shape and helps transport cargo. Key motor and scaffold proteins were reduced, while some neurofilament components accumulated, hinting at traffic jams and possible clumping. Electrical recordings from hippocampal slices confirmed that mice expressing PKN207 had weakened long‑term potentiation—the process by which synapses strengthen after repeated activity and a widely accepted cellular basis for learning and memory. Even though PKN207 lacks the enzyme domain of PKN1, its presence was enough to imitate and sometimes exceed the disruptive effects of the full protein, implying that the shared N‑terminal region can, on its own, interfere with neuronal homeostasis.
Why this discovery matters for brain disease
This work adds a new layer to our understanding of TDP-43‑related disorders. Rather than merely causing loss of essential RNAs, TDP-43 failure can also spawn a stable, toxic micro‑protein that undermines synapses and cognition. The cryptic PKN1‑5a1 exon and its peptide product PKN207 now stand out as potential biomarkers of early TDP-43 dysfunction and as candidate targets for therapies that correct splicing or block the harmful fragment. More broadly, the study suggests that other hidden exons may likewise be giving rise to disease‑driving peptides, pointing researchers toward a rich—and previously overlooked—landscape of molecular culprits in neurodegeneration.
Citation: Yang, M., Wang, Q., Yan, R. et al. A neurotoxic cryptic peptide arising from TDP-43-dependent cryptic splicing of PKN1. Nat Commun 17, 2963 (2026). https://doi.org/10.1038/s41467-026-68916-0
Keywords: TDP-43, cryptic splicing, PKN1, neurodegeneration, Alzheimer’s and ALS