Clear Sky Science · en
Human iPSC-based Modeling of Pulmonary Fibrosis Reveals p300/CBP Inhibition Suppresses Alveolar Transitional Cell State
Why scarring lungs matter to all of us
Idiopathic pulmonary fibrosis (IPF) is a relentless disease in which the lungs slowly turn into scar tissue, making every breath harder. Current drugs can only slow this process and often cause troubling side effects. This study uses cutting‑edge stem cell and genomic tools to recreate scarred lungs in the lab, asking a simple but vital question: can we find a switch that steers damaged lung cells away from a harmful state and back toward healing?
A lab-grown window into a scarred lung
To probe IPF, the researchers built miniaturized lungs from human induced pluripotent stem cells (iPSCs). These iPSCs were guided to become alveolar cells—the cells that line the tiny air sacs where oxygen enters the blood—and were grown together with lung fibroblasts, the connective tissue cells that form scar. Embedded in a soft gel, these “alveolar organoids” behaved much like real lung tissue. When exposed to the chemotherapy drug bleomycin, a known trigger of lung injury, the gels shrank as fibroblasts pulled on them, mimicking the tissue contraction seen in fibrosis.
Using this system, the team screened a library of 264 small molecules and automatically measured how much each drug prevented gel contraction, with a deep-learning image analysis tool ensuring objective readouts. Many compounds had no effect, but one family clearly stood out: inhibitors of the proteins p300 and CBP, which help control how DNA is packaged and which genes are turned on. All eight p300/CBP‑targeting compounds in the library reduced contraction at low doses, highlighting this pathway as a promising handle on fibrosis. 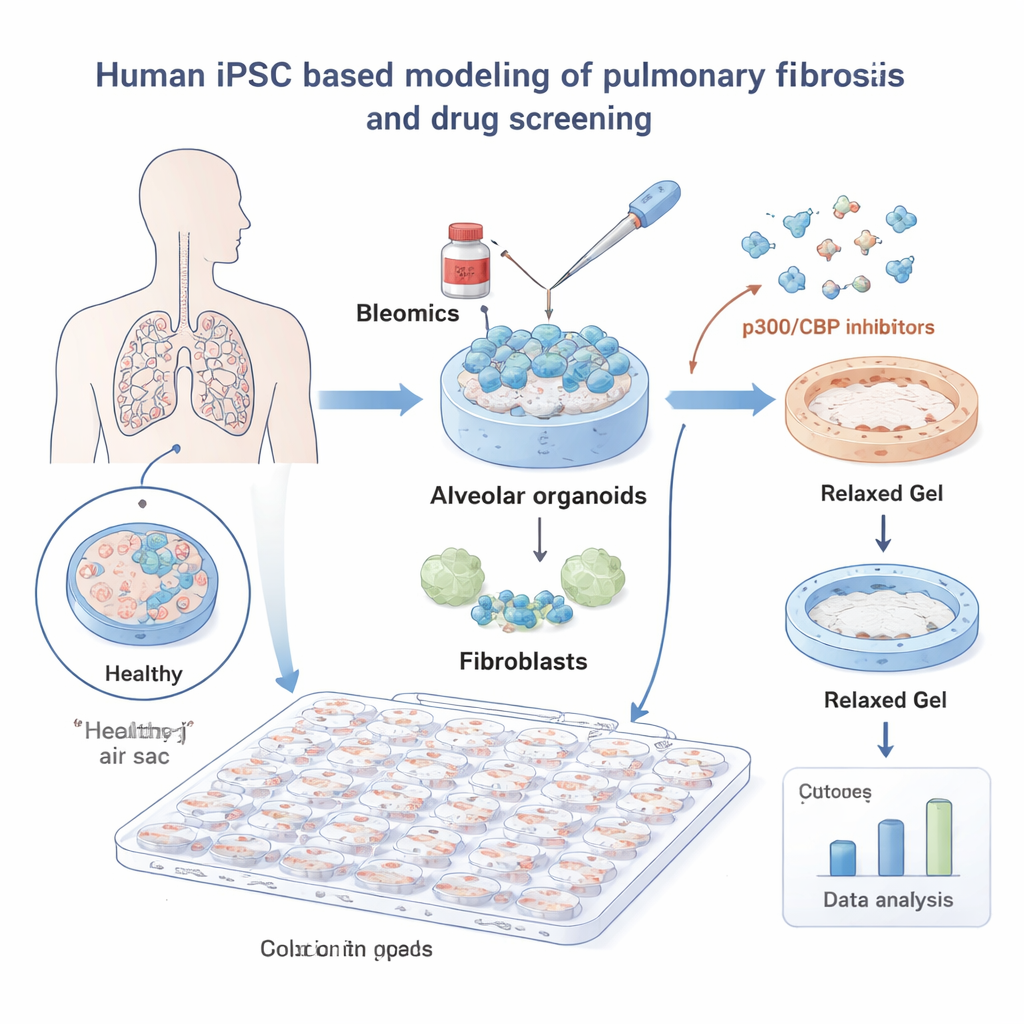
The troublemakers: transitional lung cells
Recent work has revealed a problematic “in‑between” cell type in diseased lungs, called the alveolar transitional cell state. Normally, support cells known as AT2 cells mature into ultra‑thin AT1 cells that cover the air sacs and enable gas exchange. In IPF, however, AT2 cells often stall in this transitional state, expressing stress and repair genes while failing to complete the journey to fully functional AT1 cells. These transitional cells cluster in fibrotic regions and communicate strongly with fibroblasts, but it was unclear whether they were simply a byproduct of damage or active drivers of scarring.
By sequencing RNA and profiling open chromatin in their organoids, the authors showed that the transitional cells arising in their model closely matched those found in lungs from IPF patients. These induced transitional cells displayed gene signatures of stress, inflammation, and matrix remodeling, and they strongly activated co‑cultured adult lung fibroblasts. Crucially, when p300/CBP was blocked, markers of the transitional state dropped, AT2 identity was better preserved, and fibroblast activation waned. In other words, the drugs did not broadly poison cells; instead, they selectively prevented AT2 cells from becoming trapped in this harmful limbo.
Unraveling the molecular switches
To understand how p300/CBP shapes this fate decision, the team examined chemical marks on histones—proteins that help package DNA. A particular mark, H3K27 acetylation, is commonly laid down by p300/CBP at active enhancers and promoters. In transitional cells, regions near stress‑response and pro‑fibrotic genes carried strong H3K27 acetylation and were enriched for binding sites of transcription factors such as AP‑1 and HNF1B. When cells were treated with p300/CBP inhibitors, these acetyl marks diminished at those sites, and expression of many pro‑fibrotic genes fell. Blocking AP‑1 directly, or reducing AP‑1 and HNF1B with small interfering RNAs, likewise curtailed the transitional program and organoid contraction, tying this trio—p300/CBP, AP‑1, and HNF1B—to the engine that fuels fibrotic remodeling.
Beyond the dish, the study tested one inhibitor, CBP30, in mice with bleomycin‑induced lung injury. Animals given CBP30 had fewer transitional epithelial cells, less activation of scar‑forming myofibroblasts, and reduced expression of fibrosis markers. This cross‑validation between human stem‑cell models and an animal model strengthens the case that p300/CBP is not just a lab artifact but a genuine regulator of lung scarring. 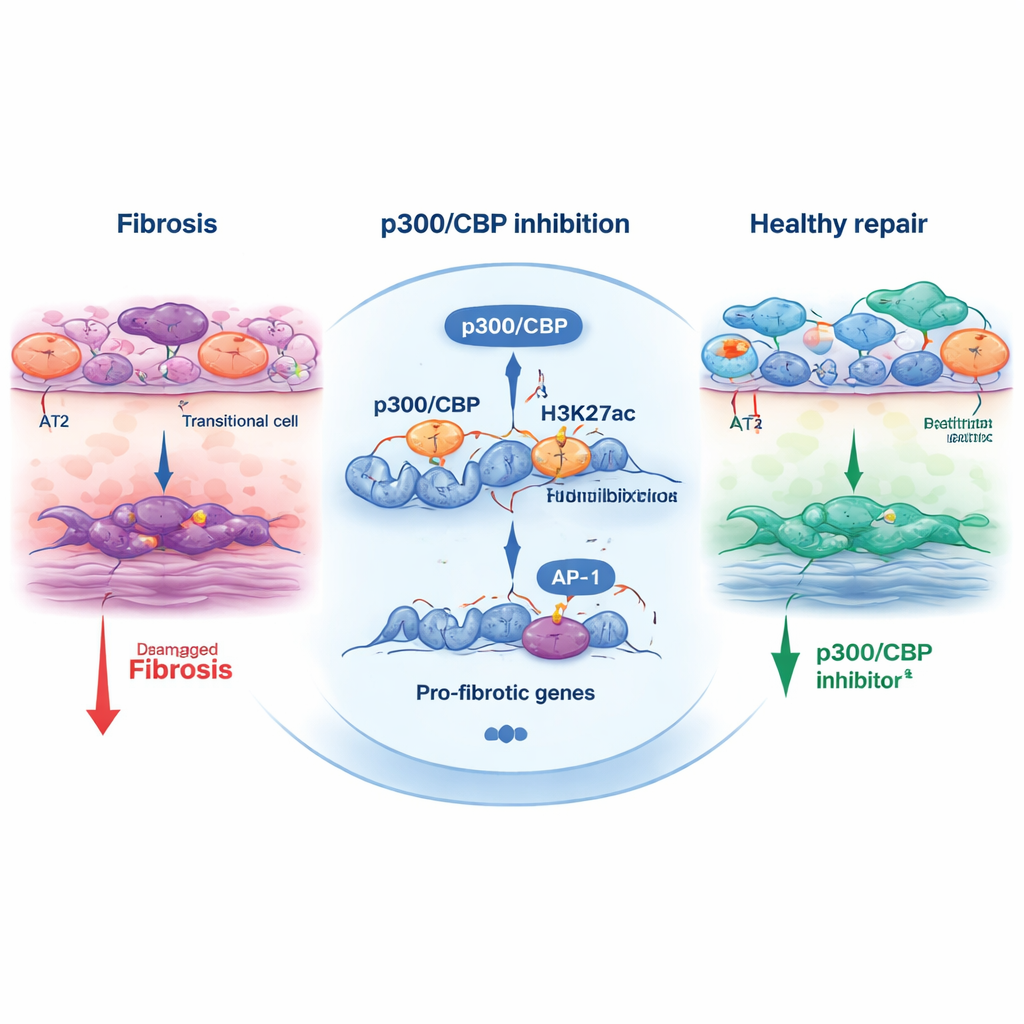
What this means for future treatments
For non‑specialists, the key takeaway is that the authors have built a realistic human model of fibrotic lungs and used it to spotlight a new drug target. Their work suggests that lung scarring is driven in part by a reversible, stress‑induced transitional cell state that misguides the surrounding tissue. By dialing down p300/CBP, it may be possible to quiet this state, keep alveolar cells on a healthy developmental track, and reduce the signals that push fibroblasts into overdrive. While p300/CBP inhibitors still need to be optimized for safety and tested clinically, this study points toward therapies that address the root cellular miscommunication in IPF rather than merely slowing its aftermath.
Citation: Tsutsui, Y., Masui, A., Konishi, S. et al. Human iPSC-based Modeling of Pulmonary Fibrosis Reveals p300/CBP Inhibition Suppresses Alveolar Transitional Cell State. Nat Commun 17, 1214 (2026). https://doi.org/10.1038/s41467-026-68909-z
Keywords: idiopathic pulmonary fibrosis, alveolar organoids, p300/CBP inhibitors, transitional epithelial cells, lung stem cells