Clear Sky Science · en
Cytoplasmic NAD/H synthesis via NRK1 regulates inflammatory capacity and promotes survival of CD4+ T cells
Keeping Immune Cells in Balance
When our bodies face infections, certain white blood cells called CD4+ T cells leap into action, helping to coordinate the immune response. But if these cells become too aggressive, they can damage our own tissues; if they are too weak, infections take hold. This study explores how a small metabolic switch inside T cells, centered on a molecule called NAD and an enzyme named NRK1, helps decide whether these cells respond in a controlled way or tip into harmful overdrive.
Fuel for Busy Immune Cells
As CD4+ T cells become activated during infection, their energy demands skyrocket. They burn more sugar, use their mitochondria more intensely, and generate bursts of reactive oxygen species (ROS)—highly reactive molecules that can act as signals but also cause damage. All of this depends on NAD, a tiny cofactor that ferries electrons around and is constantly used and recycled. The authors found that, in both human and mouse CD4+ T cells, activation strongly increases levels of the enzyme NRK1, which helps rebuild cellular NAD from precursor molecules. Adding an NAD precursor called nicotinamide riboside (NR) boosted NAD levels in human T cells, but unexpectedly made them less activated and less likely to release inflammatory messenger proteins.
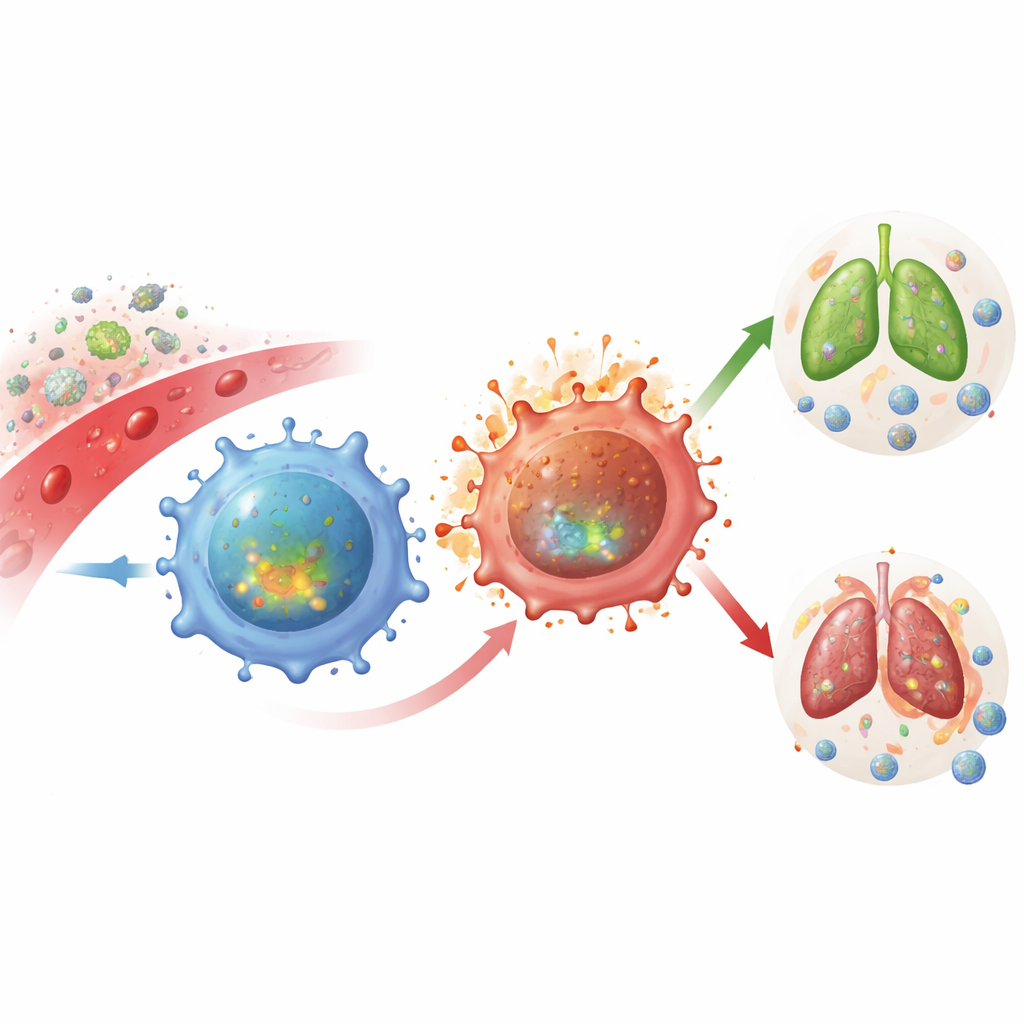
More Firepower but Shorter Life
To understand what NRK1 really does inside T cells, the team turned to mice genetically engineered to lack NRK1. Their CD4+ T cells contained less NAD overall and could no longer respond to NR. When these cells were stimulated, they actually produced more inflammatory cytokines such as interferon-gamma and other signaling molecules, suggesting they had become hyperactive. However, there was a catch: these same NRK1-deficient cells died off more readily during prolonged activation. In other words, losing NRK1 shifted T cells toward a more explosive but less sustainable response, with stronger short-term firing but reduced long-term survival.
A Redox Safety Valve Inside the Cell
The researchers then asked why altering NRK1 would change T-cell behavior so dramatically. They discovered that NRK1 is especially important for generating not only NAD, but also its phosphorylated cousin, NADP, and its reduced form, NADPH, inside the cell’s fluid interior, the cytoplasm. NADPH is a key player in antioxidant systems that recycle glutathione, one of the cell’s main defenses against ROS. In NRK1-deficient cells, NADP/NADPH levels fell more sharply than NAD itself, glutathione defenses weakened, ROS levels rose, and a transcription factor called NFAT was more likely to move into the nucleus and turn on inflammatory genes. Blocking a separate enzyme that makes NADPH reproduced this rise in ROS and cytokine output, while treating cells with an antioxidant reversed the hyper-inflammatory state. In human T cells, providing NR increased NADPH, strengthened antioxidant capacity, reduced ROS, and kept NFAT out of the nucleus, again dampening inflammation.
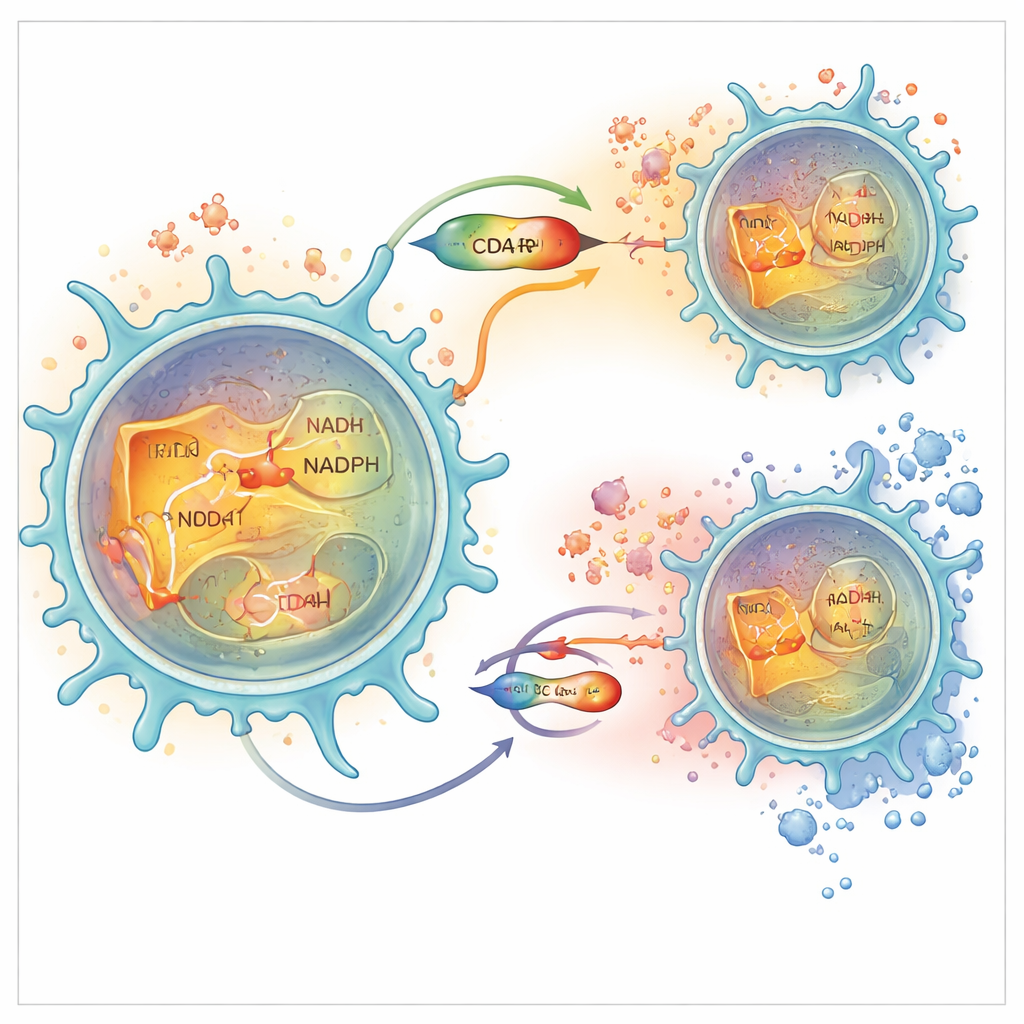
Local Control in the T Cell Interior
Digging deeper, the team showed that NRK1 levels rise mainly in the cytoplasm of activated CD4+ T cells, not in their mitochondria, and that partner enzymes there are tuned to convert NR-derived intermediates into NAD and then NADP/NADPH. Using both fluorescent biosensors and biochemical fractionation, they verified that NRK1 activity locally boosts NAD and NADPH specifically in this compartment. This local "metabolic pocket" is closely linked to glycolysis, the sugar-burning pathway in the cytoplasm, and to ROS handling. Without NRK1, cells shifted away from glycolysis toward heavier use of mitochondrial oxidation but did not show massive energy failure, indicating that the main consequence of losing NRK1 is disruption of redox balance and signaling rather than wholesale shutdown of metabolism.
Real-World Tests in Infection
To see how this mechanism plays out in living animals, the researchers studied mice whose T cells alone lacked NRK1 during serious infections with a lung fungus (Cryptococcus neoformans) and influenza virus. In both cases, NRK1-deficient CD4+ T cells showed signs of greater DNA damage—likely driven by uncontrolled ROS—and were less able to persist as functional effector cells at key sites such as the brain during fungal infection and lymph nodes draining infected lungs in flu. Mice with NRK1-deficient T cells had higher fungal loads in the brain and worse disease scores during influenza, tying the biochemical pathway directly to the ability to control pathogens.
What This Means for Future Therapies
Overall, the study reveals that NRK1 acts as a crucial internal moderator for CD4+ T cells, shaping both how strongly they inflame tissues and how long they survive. By steering cytoplasmic production of NAD and NADPH, NRK1 supports antioxidant defenses, restrains excessive inflammatory signaling, and helps maintain effective T-cell numbers during infection. For a lay reader, the message is that the immune system’s power and precision depend not only on which cells are present, but also on tiny metabolic circuits within those cells. Adjusting NAD-related pathways—for example with supplements like nicotinamide riboside or drugs targeting NRK1 and its partners—could one day offer new ways to calm damaging inflammation or bolster immune defense, depending on clinical need.
Citation: Stavrou, V., Ali, M., Gudgeon, N. et al. Cytoplasmic NAD/H synthesis via NRK1 regulates inflammatory capacity and promotes survival of CD4+ T cells. Nat Commun 17, 2349 (2026). https://doi.org/10.1038/s41467-026-68863-w
Keywords: CD4 T cells, NAD metabolism, oxidative stress, immune regulation, nicotinamide riboside