Clear Sky Science · en
Evaluating single-cell ATAC-seq atlasing technologies using sequence-to-function modeling
Reading the cell’s instruction manual
Every cell in your body reads the same DNA, yet brain cells, muscle cells, and immune cells behave very differently. This paper tackles a core puzzle behind that diversity: how short stretches of DNA called enhancers act like switches to turn genes on and off in specific cell types. The authors show that new, cheaper lab technology can generate the massive datasets needed to train modern deep‑learning models that read DNA sequences and predict which enhancers are active in which cells, bringing us closer to truly decoding the genome’s regulatory “grammar.”
Making maps of open DNA in single cells
Enhancers usually sit in stretches of DNA that are more open and accessible, making them easier for regulatory proteins to bind. A technique called single‑cell ATAC‑seq measures which parts of the genome are open in thousands to hundreds of thousands of individual cells at once, creating an “atlas” of accessible DNA across many cell types. These atlases are ideal fuel for deep‑learning models that take raw DNA sequence as input and learn to predict how strongly each small region acts as an enhancer in each cell type. Until now, however, most such atlases have relied on expensive commercial instruments, raising the question of whether low‑cost, open‑source methods can provide training data of equal value for these models.
An open-source alternative to commercial platforms
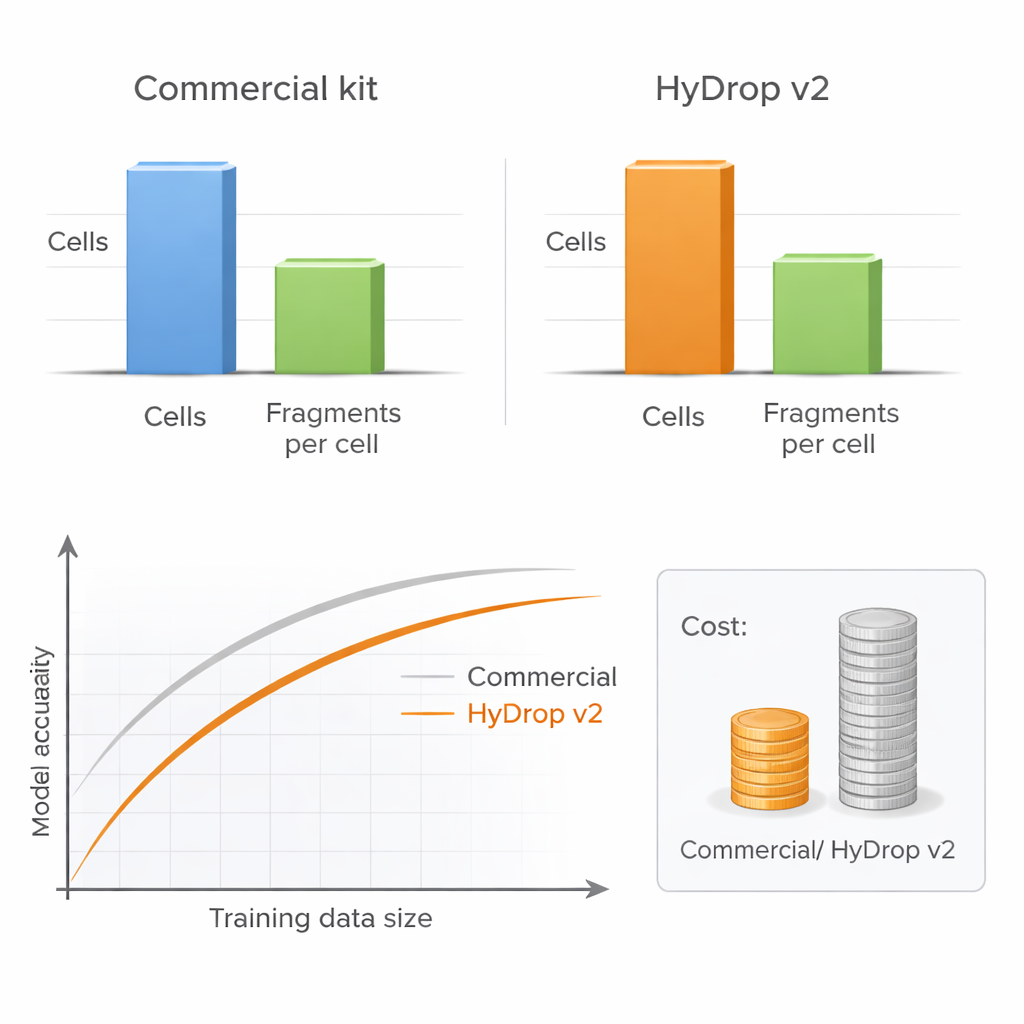
The authors introduce HyDrop v2, an improved droplet‑based method for single‑cell ATAC‑seq that uses custom hydrogel beads to barcode individual cells. They benchmark HyDrop v2 against a widely used commercial kit by building large atlases from two very different systems: the adult mouse motor cortex and late‑stage fruit fly embryos. HyDrop v2 generates comparable data quality—recovering the same major cell types and very similar sets of accessible DNA regions—while costing about fourteen times less per mouse brain sample. Importantly, data from HyDrop v2 experiments integrate smoothly with commercial data, meaning researchers can mix and match platforms when building very large atlases.
Training deep-learning models to read enhancer logic
To test whether cheaper data are good enough for advanced modeling, the team trains sequence‑to‑function deep‑learning models on either commercial or HyDrop v2 atlases. These models learn directly from DNA sequence to predict how accessible each region is in each cell type, and can highlight short sequence patterns that likely correspond to binding sites for specific regulatory proteins. In the mouse cortex, models trained on HyDrop v2 data match commercial‑data models in overall accuracy and in their ability to recover known enhancer “switches” that were previously validated in live animals. In the fly embryo, both platforms support models that can zoom into 2,000‑base‑pair regions and pinpoint the core ~500‑base‑pair segments that actually drive tissue‑specific enhancer activity, such as regions that control neuroblast or muscle gene expression.
More cells can beat more depth
A key practical question for any lab is whether to sequence each cell very deeply or to profile more cells at lower depth. By systematically varying the number of cells and the number of DNA fragments per cell, the authors show that model performance hardly suffers when sequencing depth is reduced to a moderate level, as long as enough cells are included. In contrast, cutting the number of cells clearly harms model accuracy, especially when measuring performance across many cell types at once. Because HyDrop v2 is much cheaper per cell, researchers can readily add tens of thousands of extra cells, recovering or even surpassing the performance of commercial‑based models at a fraction of the cost.
Seeing protein footprints on DNA
The study also examines whether different lab platforms introduce subtle biases into how the ATAC‑seq enzyme cuts DNA, which could mislead models that try to infer where proteins sit on the genome. Using a separate neural‑network tool that corrects for enzyme preferences, the authors show that HyDrop v2 and commercial kits produce nearly identical patterns of enzyme activity in both mouse and fly cells. After correction, both datasets reveal fine‑scale “footprints” where regulatory proteins and nucleosomes appear to protect DNA from cutting, and these footprints line up with the sequence patterns highlighted by the sequence‑to‑function models. This agreement suggests that open‑source and commercial platforms are equally suitable for detailed studies of how proteins interact with DNA.
Why this matters for decoding the genome
For non‑specialists, the take‑home message is that we can now build very large, affordable maps of how DNA is used in individual cells, and train powerful deep‑learning models on those maps without relying solely on costly proprietary hardware. HyDrop v2 delivers data that support enhancer prediction, interpretation of sequence patterns, and protein‑binding footprints on par with leading commercial methods, provided enough cells are profiled. This opens the door to constructing organism‑wide atlases of regulatory elements in health and disease, accelerating efforts to read the genome’s regulatory instructions and to design new, precisely targeted genetic switches for research and future therapies.
Citation: Dickmänken, H., Wojno, M., Mahieu, L. et al. Evaluating single-cell ATAC-seq atlasing technologies using sequence-to-function modeling. Nat Commun 17, 1951 (2026). https://doi.org/10.1038/s41467-026-68742-4
Keywords: single-cell ATAC-seq, enhancers, deep learning models, gene regulation, open-source genomics