Clear Sky Science · en
Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer
Why predicting enzyme jobs matters
Every living cell runs on countless tiny chemical machines called enzymes. Each enzyme has a specific “job,” and that job is encoded in an Enzyme Commission (EC) number, a four-part code a bit like a postal address. Correctly assigning EC numbers is crucial for understanding metabolism, designing new drugs, engineering microbes to make fuels or plastics alternatives, and tracking how ecosystems process chemicals. But experiments to determine enzyme functions are slow and expensive. This study introduces HIT-EC, a new artificial intelligence model that can reliably predict EC numbers from protein sequences while also explaining why it made each prediction.
A zip code system for enzyme jobs
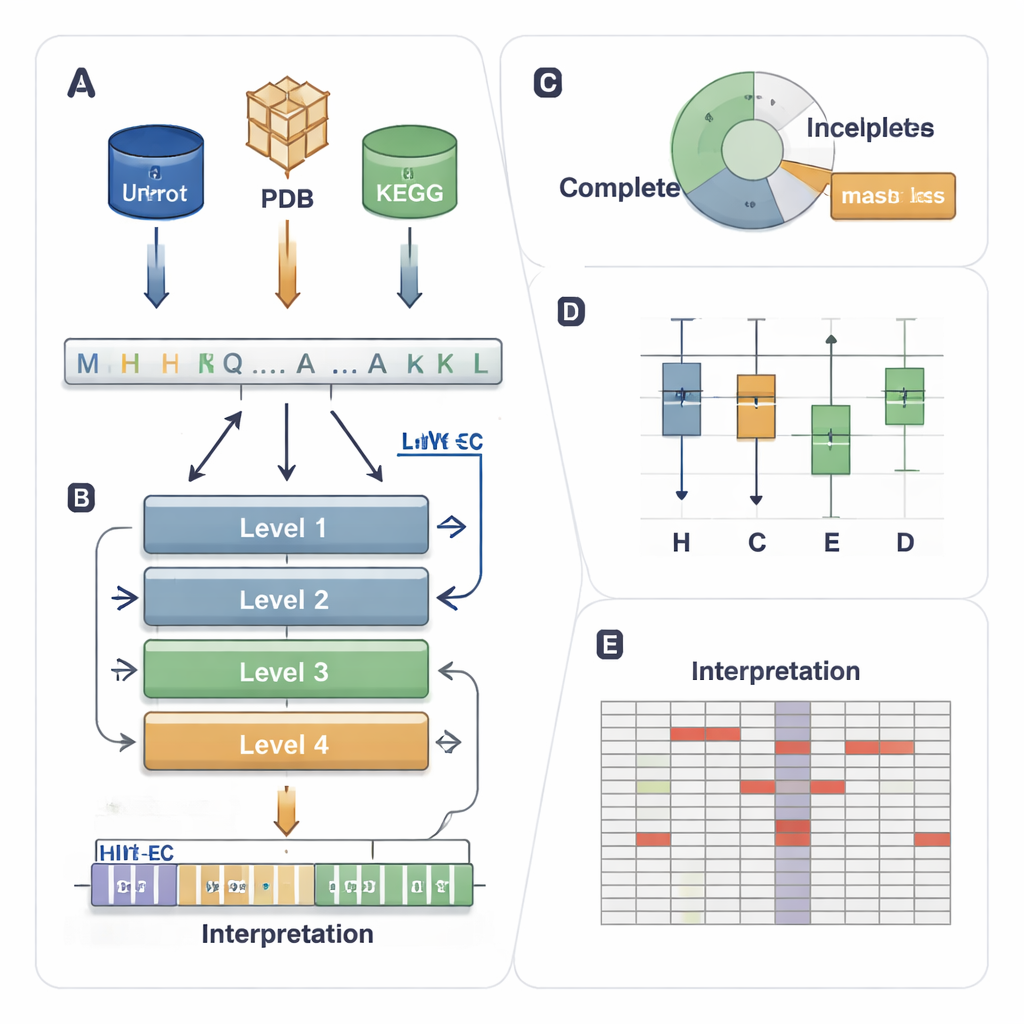
The EC system assigns each enzyme a four-level code such as 1.1.1.37. The first digit indicates a broad class (for example, enzymes that move electrons or transfer groups), and the later digits describe more precise reaction details. This hierarchy is powerful but creates a demanding prediction problem: a model must get all four levels right for thousands of possible codes, even when some enzymes are rare or only partly annotated in databases (for example, 3.5.-.-, where the detailed levels are missing). Existing computer methods use either 3D structure, sequence similarity, or deep learning, but they tend to struggle with uncommon enzymes, ignore partially labeled data, and typically behave as “black boxes” that offer little insight into why they made a call.
A four-story AI that follows the EC ladder
HIT-EC (Hierarchical Interpretable Transformer for EC prediction) is built to mirror the four-step EC hierarchy. It takes a raw protein sequence and passes it through four transformer layers, each focused on one EC level. Local flows connect each level to the previous one, ensuring that a fine-grained decision (the fourth digit) must be consistent with broader ones (the first and second digits). In parallel, a global flow keeps the full sequence context visible at each step. The model can also be trained on sequences with incomplete labels, using a “masked loss” that simply ignores missing EC levels instead of throwing the sequence away. This lets HIT-EC learn from the large fraction of proteins in curated databases that are only partly annotated.
Outperforming rivals on accuracy and speed
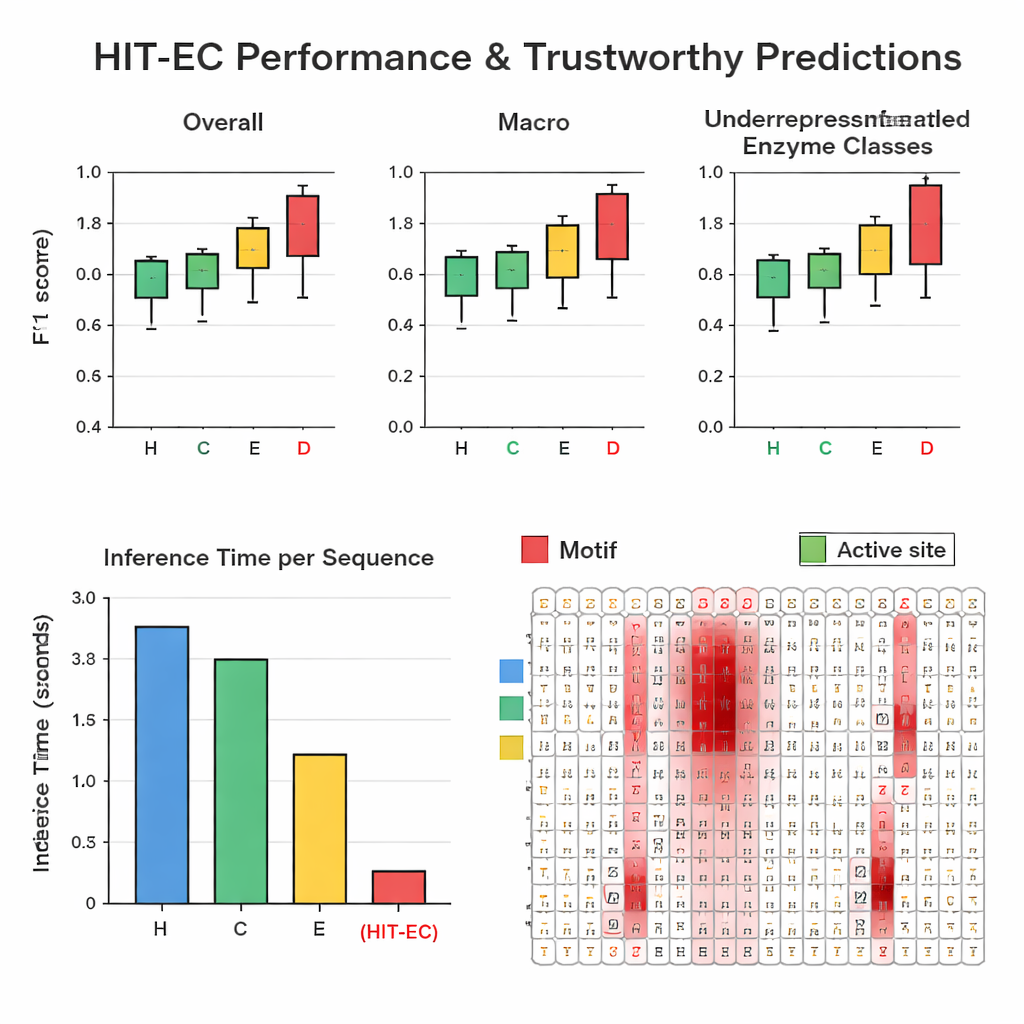
The authors assembled a large, carefully filtered dataset of about 200,000 enzymes with 1,938 different EC numbers from Swiss-Prot and the Protein Data Bank. In repeated hold-out tests, HIT-EC beat three leading methods (CLEAN, ECPICK, and DeepECtransformer) on both overall and per-class F1-scores, which measure the balance between correct hits and false alarms. It was particularly strong on underrepresented EC codes with 25 or fewer known examples, where earlier methods often falter. HIT-EC also generalized well to new enzymes added to Swiss-Prot after training and to full genomes from diverse bacteria, including well-studied strains of Escherichia coli, Bacillus subtilis, and Mycobacterium tuberculosis. Despite its sophistication, the model was highly efficient: on a standard GPU it processed a protein in about 38 milliseconds—tens of times faster than some competitors that depend on slower similarity searches or ensembles of many models.
Seeing what the model “looks at”
To make its predictions trustworthy, HIT-EC is designed to show which amino acids in the sequence influenced each EC-level decision. The authors built an interpretation pathway that combines attention weights with gradient information to score how important each position is. They validated these scores on well-characterized enzyme families. For example, in a cytochrome P450 family (CYP106A2), HIT-EC highlighted known functional motifs such as oxygen-binding and heme-binding regions, and identified a subtle EXXR motif that one benchmark model missed. For classic representatives of each top-level EC class—such as alcohol dehydrogenase, hexokinase, and carbonic anhydrase—the model’s relevance scores lit up textbook signature motifs and substrate-binding sites. These interpretations provide biochemical “evidence” that the model is basing its calls on meaningful features, not on accidental correlations.
Guiding work on rare and emerging enzymes
The team further tested HIT-EC on two understudied enzymes important for pollution cleanup: a cytochrome P450 involved in breaking down aromatic pollutants, and a PET-degrading hydrolase from Streptomyces that helps digest plastic-related molecules. Both enzymes had been experimentally characterized but lacked official EC assignments. HIT-EC correctly predicted the expected EC numbers and highlighted motif patterns and catalytic residues that match what is known from structural and biochemical studies. Overall, the work shows that HIT-EC can not only assign EC numbers more accurately and quickly than current tools, especially for rare functions, but also shed light on why a particular enzyme is believed to perform a given chemical task. This blend of performance and interpretability makes it a promising engine for large-scale, reliable enzyme annotation in genomics, biotechnology, and environmental research.
Citation: Dumontet, L., Han, SR., Lee, J.H. et al. Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer. Nat Commun 17, 1146 (2026). https://doi.org/10.1038/s41467-026-68727-3
Keywords: enzyme function prediction, deep learning in biology, transformer models, protein annotation, bioremediation enzymes