Clear Sky Science · en
An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression
When Cell Antennas Go Rogue
Why do some cancers keep growing even after rounds of chemotherapy and cutting-edge immunotherapy? This study follows a patient with tumors in both the lung and brain and traces their disease back to a tiny change in a key cell-surface "antenna" called EGFR. By uncovering how this single mutation rewires growth signals, the researchers not only explain the patient’s aggressive cancer, but also show how an existing drug, afatinib, can rein it back in.
A Rare Mutation With Big Consequences
EGFR is a receptor that sits across the cell membrane and helps cells respond to growth cues. Many lung and brain cancers carry mutations in EGFR, but most known alterations sit inside the cell, in the portion that acts like an enzyme switch. Here, the team discovered an unusual change on the outside of EGFR, in the part that normally grabs growth factors. In a patient with both lung cancer and glioma, they found that one amino-acid building block at position 252 was swapped from arginine to cysteine—dubbed EGFR R252C. Mining cancer databases showed this mutation in a small fraction of glioma patients and almost never in lung tumors, hinting it is rare but real. Using gene-editing tools, the authors recreated this exact mutation in several human brain and lung cancer cell lines to test what it does.
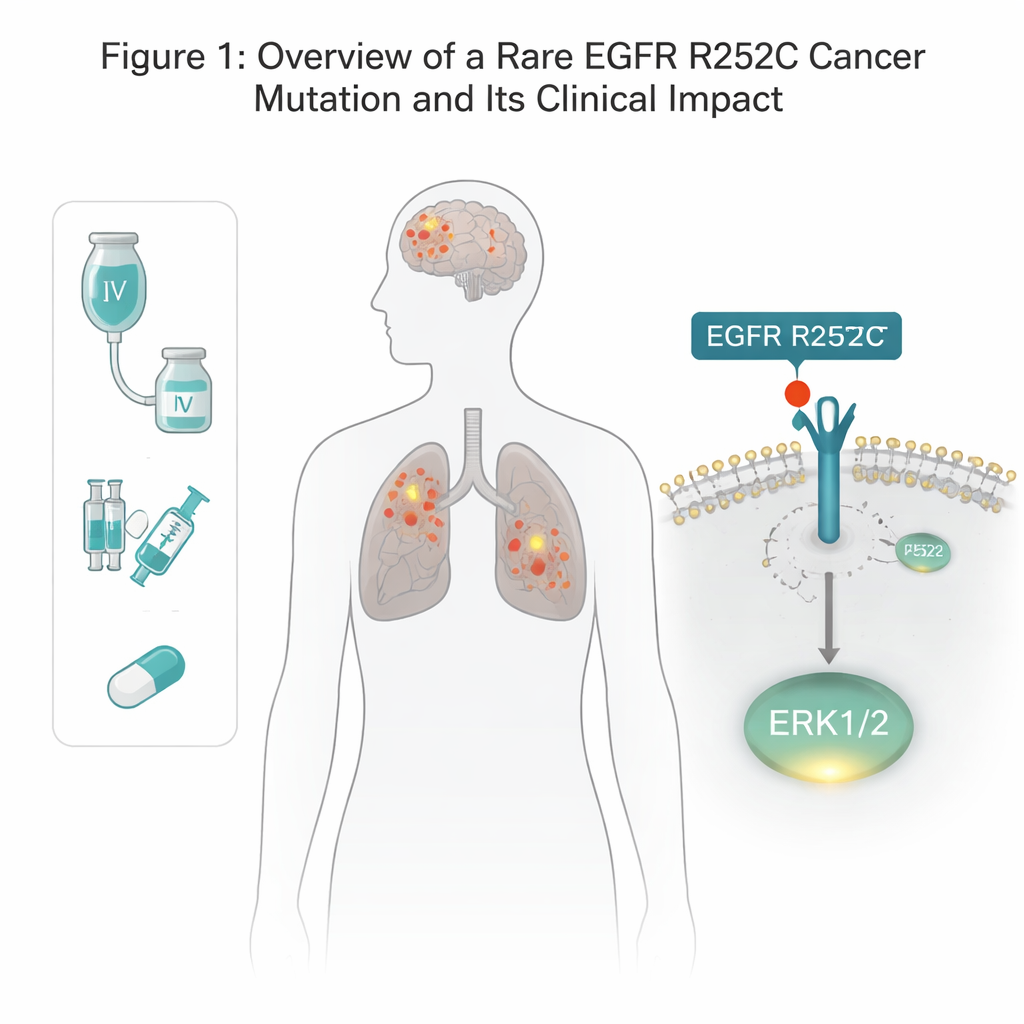
A New Shortcut to Growth Signals
Ordinarily, EGFR must pair up with another copy and then tag its own inner tail with phosphate groups before it can switch on downstream growth pathways. Surprisingly, the R252C version did not show this usual self-tagging. Instead, cells carrying EGFR R252C turned on one specific growth controller, ERK1/2, far more strongly than normal, while leaving other classic EGFR routes—such as AKT and STAT3—largely unchanged. Blocking ERK1/2 with a dedicated inhibitor erased the extra growth advantage of R252C cells, proving that ERK1/2 is the main engine behind this mutant’s tumor-driving power.
Locking the Receptor in an Always-On Shape
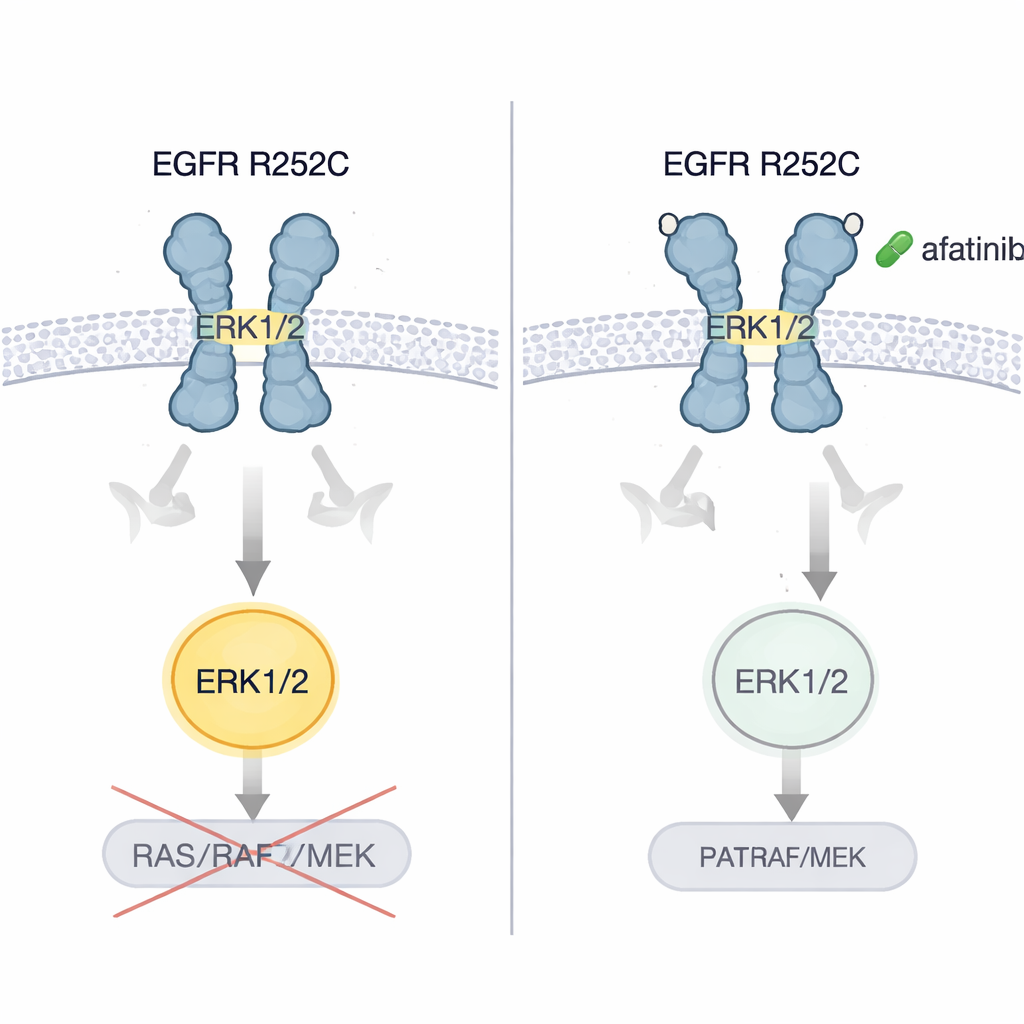
To understand how an external change could cause such selective overdrive, the researchers combined biochemical assays with computer simulations. The R252C swap introduces a new cysteine on the outer portion of EGFR. Two such mutants can form a disulfide bond—a kind of molecular staple—between their C252 residues, locking them together into a stable pair. Structural modeling showed that this bond forces the outside of the receptor into a “V-shaped,” staggered alignment that closely mimics the active, ligand-bound form, even without any growth factor present. This alignment propagates through the membrane-spanning and just-inside segments, twisting the internal enzyme domains into an unusual arrangement: the active sites face into the cell but are held too far apart to efficiently tag each other. Instead, this conformation creates a strong docking surface for ERK1/2, allowing EGFR R252C to directly phosphorylate ERK1/2 and bypass the usual RAS–RAF–MEK relay.
From Mouse Models to a Single Patient
The authors showed that brain and lung cancer cells carrying EGFR R252C grew faster in dishes and formed larger, more aggressive tumors when implanted into mice, compared with cells carrying normal EGFR. They then tested several generations of EGFR-blocking pills. Only afatinib, a second-generation inhibitor, consistently shut down ERK1/2 activation and sharply reduced tumor cell growth. In mouse models of brain and lung tumors driven by R252C, afatinib slowed tumor expansion and extended survival. Critically, when the original patient—whose disease had worsened despite chemotherapy, a blood-vessel-targeting drug, and immunotherapy—was switched to afatinib, scans of both lung and brain showed marked shrinkage of tumor burden and the patient enjoyed several years without progression.
What This Means for Patients
This work reveals a previously unrecognized way that a cancer-causing EGFR mutation can operate: by stapling two receptors together outside the cell, twisting them into an active pose that directly flips on ERK1/2 rather than following the textbook signaling chain. For non-specialists, the key takeaway is that not all mutations in the same gene behave alike, and some rare changes may respond best to specific existing drugs. EGFR R252C appears to create cancers that are particularly vulnerable to afatinib. While this conclusion currently rests on one detailed patient case plus extensive lab work, it points toward more personalized testing of EGFR’s outer-domain mutations and suggests that carefully chosen targeted therapies could offer new hope to select patients with difficult-to-treat brain and lung tumors.
Citation: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Keywords: EGFR mutation, glioma, lung cancer, ERK signaling, afatinib