Clear Sky Science · en
Improving polygenic score prediction for underrepresented groups through transfer learning
Why your DNA risk score may not work for you
Genetic "risk scores" are increasingly used to estimate a person’s chance of developing common conditions such as diabetes, heart disease, or high blood pressure. But most of these scores were built using DNA data from people of European ancestry. As a result, they often predict poorly for people from other backgrounds, raising concerns about fairness and usefulness in real-world medicine. This study asks a simple question: can we reuse what we’ve learned from large European datasets to build better, fairer genetic scores for underrepresented groups—without sharing anyone’s raw data?
From millions of DNA markers to a single risk score
A polygenic score is like a report card that adds up the small effects of many genetic markers spread across the genome. Each marker gets a weight that reflects how strongly it is associated with a trait, based on large genetic studies. When these studies involve mostly Europeans, the resulting score tends to work best in Europeans. Differences in genetic backgrounds—how common certain DNA variants are and how they are inherited together—mean that the same weights often misfire in African American, Hispanic, and other populations. Collecting equally large datasets for every group is expensive and slow, so the authors turned to a machine-learning strategy called transfer learning: instead of starting from scratch in each population, they refine an existing model trained elsewhere.
How to borrow knowledge without sharing raw data
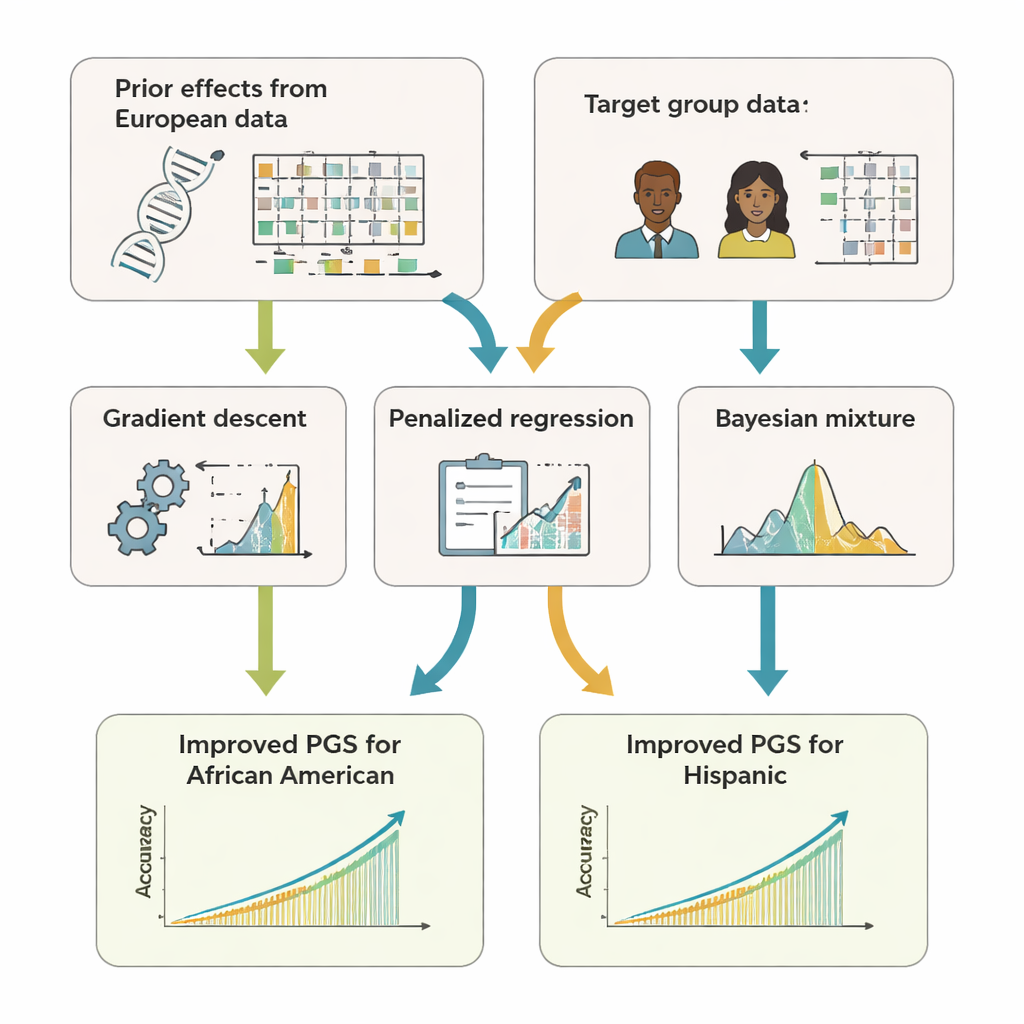
The team developed GPTL, an open-source R software package that implements three transfer-learning approaches for genetic scores. All three start from existing estimates of DNA effects obtained in a large European-ancestry dataset and then gently adjust those estimates using data from a target group, such as African Americans or Hispanics. One method tweaks the European weights step by step using gradient descent and stops early, before it completely overwrites them. A second method, called penalized regression, actively pulls the new estimates toward the original values unless the target data provide strong evidence otherwise. The third, a Bayesian mixture model, lets each DNA marker choose among several information sources—such as multiple ancestry groups or even a "no-effect" option—and blends them according to how well they explain the target data.
Putting the methods to the test
To see how well these approaches work, the authors used both computer simulations and real data from hundreds of thousands of volunteers in the UK Biobank and the U.S. All of Us research program. They focused on African American and Hispanic participants as target groups and used European-ancestry data as the main source of prior information. Across 11 traits—including height, body mass index, blood lipids, blood pressure, and kidney markers—the transfer-learning scores consistently predicted better than scores built only within the target group or simply reused from Europeans. Often, their accuracy matched or slightly exceeded that of more complex "multi-ancestry" methods that require combining raw data from several populations. Crucially, GPTL’s methods only need summary statistics—aggregated numbers about genetic effects—so institutions can collaborate without exposing individual-level genetic records.
When more DNA isn’t always better
The researchers also examined how best to choose which genetic markers to include. Contrary to the common belief that using every available marker always helps, they found that for African American and especially Hispanic groups, including millions of very weak signals could actually hurt performance, particularly when using highly simplified representations of genetic correlations. Focusing on better-supported markers and using richer information about how variants are inherited together often yielded more accurate scores. The study also showed that adding prior information from multiple ancestry groups and carefully modeling differences between populations further improved predictions.
What this means for fairer genetic risk prediction
For non-European populations, today’s off-the-shelf genetic risk scores can underperform by large margins, potentially widening health disparities. This work demonstrates that transfer learning—smartly refining existing European-based scores using modest datasets from underrepresented groups—can close much of that gap. In practice, this means that health systems and researchers can build more accurate and equitable genetic tools without pooling raw data across institutions or ancestries, easing privacy concerns. While no single method will be best for every trait and population, the GPTL toolkit shows that fairer genetic prediction is technically within reach if we treat past models not as fixed products, but as starting points that can be adapted to everyone.
Citation: Wu, H., Pérez-Rodríguez, P., Boehnke, M. et al. Improving polygenic score prediction for underrepresented groups through transfer learning. Nat Commun 17, 1973 (2026). https://doi.org/10.1038/s41467-026-68696-7
Keywords: polygenic risk scores, transfer learning, genetic prediction, health disparities, population genetics