Clear Sky Science · en
Differential membrane lipid disruption by lipopeptide antibiotics, colistin and turnercyclamycins
Why this matters for future antibiotics
Drug-resistant infections are projected to kill tens of millions of people each year by mid-century, and some of the deadliest culprits are Gram-negative bacteria that already shrug off many antibiotics. Colistin is one of the few remaining drugs that can still treat these infections, but it is harsh on patients and bacteria are increasingly learning to evade it. This study dissects how colistin works compared with a new family of natural antibiotics called turnercyclamycins, revealing that tiny chemical tweaks can produce very different ways of killing bacteria—and potentially safer medicines.
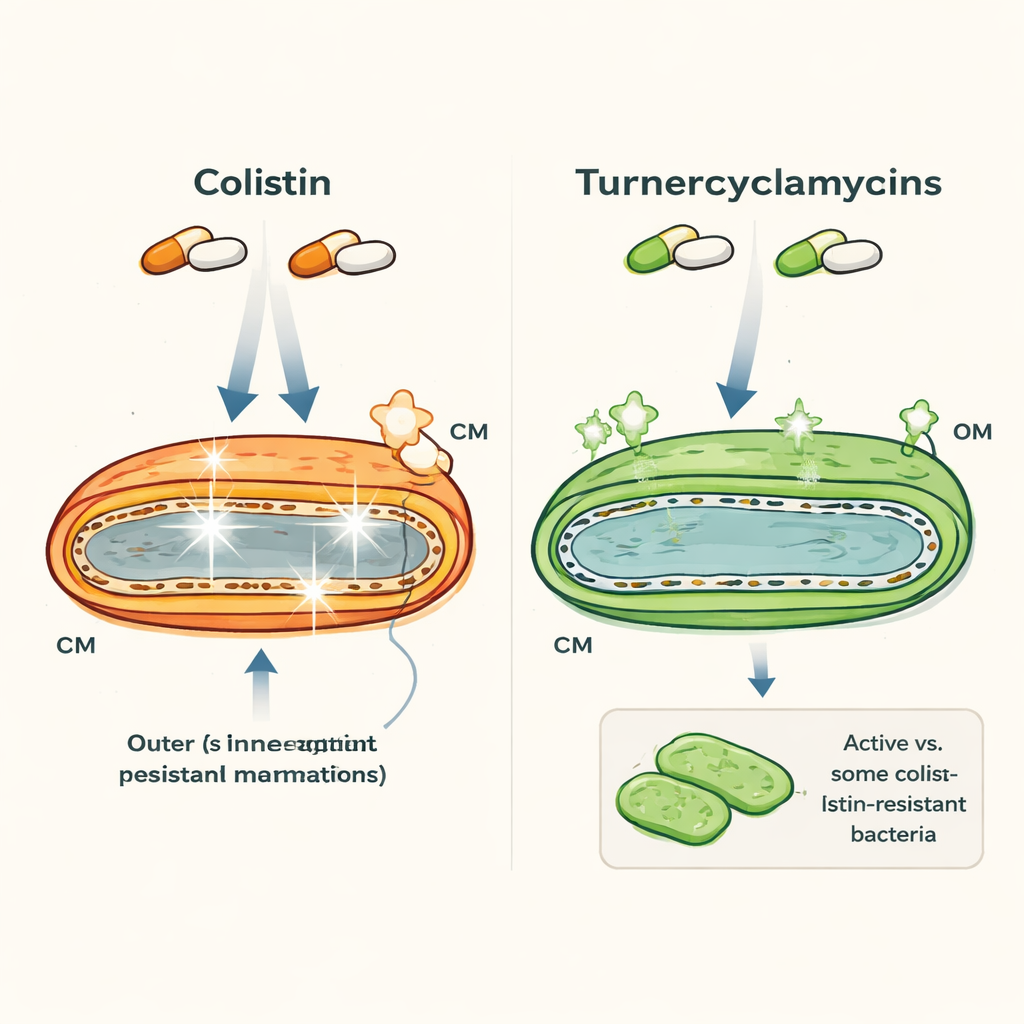
Two similar-looking drugs with very different behavior
Colistin and turnercyclamycins are both lipopeptides—molecules that combine a fatty tail with a peptide ring—and both are aimed at Gram-negative bacteria such as Escherichia coli and Acinetobacter. At first glance they look alike, yet their properties diverge sharply. Colistin is a last-resort drug that acts quickly but can damage human kidneys and nerves, and bacteria increasingly carry resistance genes such as mcr-1. Turnercyclamycins, discovered from shipworm-associated bacteria, can kill many of the same pathogens, including some colistin-resistant strains, but show far less toxicity in laboratory tests. Intriguingly, two versions that differ only by a small extension of the fatty tail already display different resistance patterns, hinting that very subtle structural features matter.
How colistin punches holes and turnercyclamycins bide their time
The authors used fluorescent dyes, time-kill experiments, and electron microscopy to watch how these drugs affect bacterial membranes over time. Colistin rapidly makes both the outer membrane and the inner, cytoplasmic membrane leaky, leading to swift cell death within a few hours. Turnercyclamycins, in contrast, kill more slowly—taking 6 to 10 hours to fully clear cultures—and mainly disturb the outer membrane. Dyes that light up when the inner membrane is breached showed strong signals for colistin but only modest, delayed signals for turnercyclamycins, and high-resolution images confirmed little visible damage to the inner membrane even when cells were dying. This indicates that turnercyclamycins do not kill by the classic “pore-forming” route used by many membrane-targeting antibiotics.
Lipid building blocks as hidden control knobs
Both drug families ultimately depend on a bacterial component called lipopolysaccharide (LPS), which is built in the inner membrane and usually exported to the outer surface. When the researchers disabled early steps of LPS biosynthesis, both colistin and turnercyclamycins lost activity; but when they blocked the transport machinery that moves LPS outward, the drugs still worked. This means that the existence of LPS building blocks is essential, but their final destination is not. A key difference emerged when binding was measured directly: colistin sticks to purified LPS with micromolar strength, while turnercyclamycins showed no measurable binding. Instead, turnercyclamycins were strongly affected by other membrane lipids. Certain phospholipids, especially phosphatidylglycerol, could blunt or modulate their activity, and the drugs were readily captured by outer membrane vesicles—tiny lipid bubbles shed by bacteria.
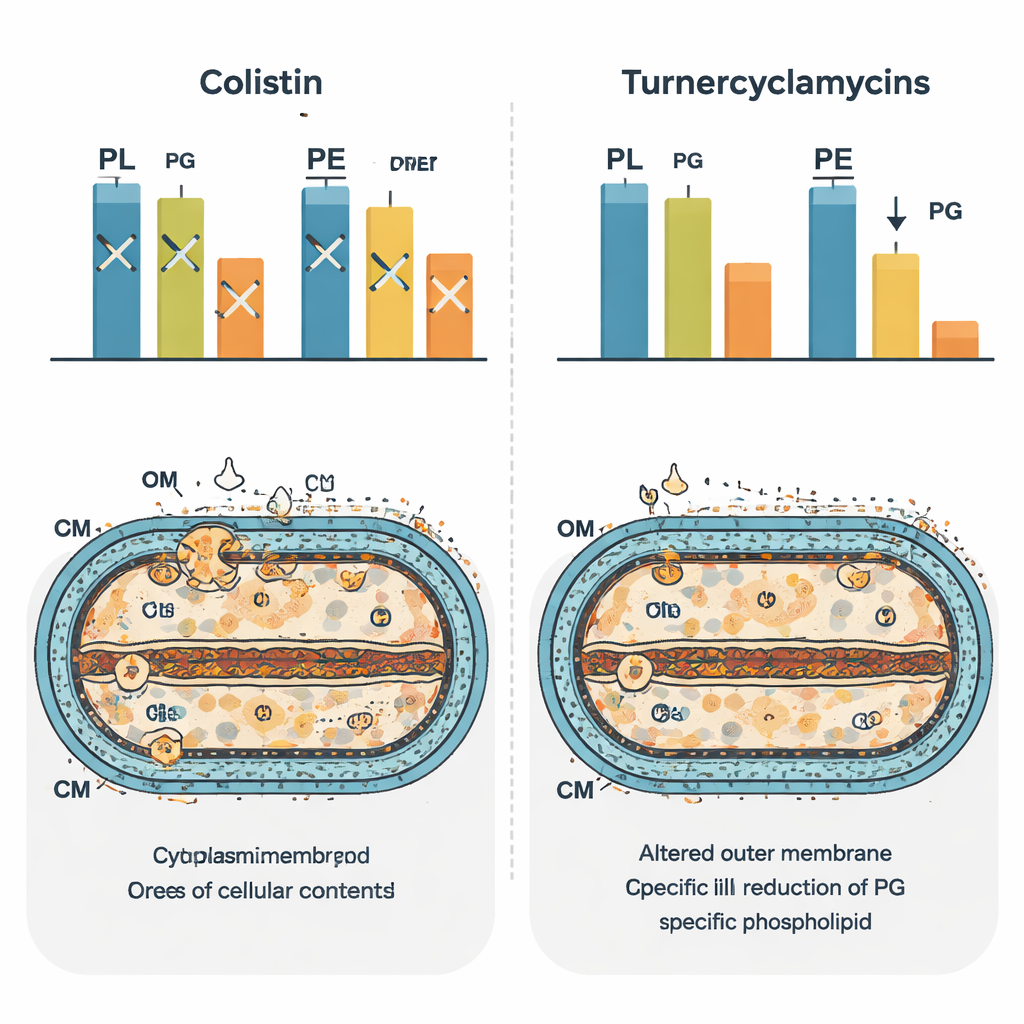
Rewiring the bacterial lipid landscape
To see the broader impact on the cell, the team used mass-spectrometry-based “phospholipidomics” to catalog hundreds of lipid species after treatment. Colistin produced one distinctive pattern of changes, reflecting its broad disruption of membrane homeostasis. Turnercyclamycins generated a different signature that closely resembled bacteria lacking a lipid transporter protein called MlaA. In these cells, certain diacyl lipids were depleted and monoacyl forms increased, suggesting that the normal cycling and remodeling of phospholipids between membranes had been thrown off balance. Notably, levels of phosphatidylglycerol dropped in turnercyclamycin-treated cells, reinforcing the idea that this lipid is directly tied to their potency. The authors propose that turnercyclamycins may act as molecular mimics that jam the pathways linking fatty-acid, LPS, and phosphatidylglycerol synthesis or transport.
What this means for designing better drugs
In plain terms, the study shows that colistin kills by rapidly tearing open both of a Gram-negative bacterium’s protective layers, helped by tight binding to LPS, whereas turnercyclamycins work more like saboteurs of the cell’s lipid supply chain. They slip into the outer membrane, gradually disturb how specific lipids are made and recycled, and eventually cause the cell envelope to fail—without heavily battering the inner membrane. Because this gentler, more targeted mechanism is linked to lower toxicity and a different resistance profile, understanding it offers a roadmap for crafting next-generation lipopeptide antibiotics. By fine-tuning features such as the length and saturation of the fatty tail, chemists may be able to build drugs that spare human tissues, retain activity against colistin-resistant strains, and stay one step ahead in the ongoing arms race with antibiotic-resistant bacteria.
Citation: Lim, A.L., Miller, B.W., Fisher, M.A. et al. Differential membrane lipid disruption by lipopeptide antibiotics, colistin and turnercyclamycins. Nat Commun 17, 1880 (2026). https://doi.org/10.1038/s41467-026-68681-0
Keywords: antibiotic resistance, Gram-negative bacteria, colistin, lipopeptide antibiotics, membrane lipids