Clear Sky Science · en
Protein folding stability estimation with explicit consideration of unfolded states
Why protein stability matters
Every protein in your body is a tiny molecular machine that must fold into a precise three‑dimensional shape to work properly. If that fold is too fragile, the protein can misfire, clump together, or fail to be produced at all—problems linked to diseases and to failures in making protein‑based drugs and enzymes. Measuring how stable a protein is in the lab is slow and tricky, so scientists are searching for computer methods that can reliably tell us, from the sequence alone, how easily a protein will unfold.
A fresh look at folded and unfolded proteins
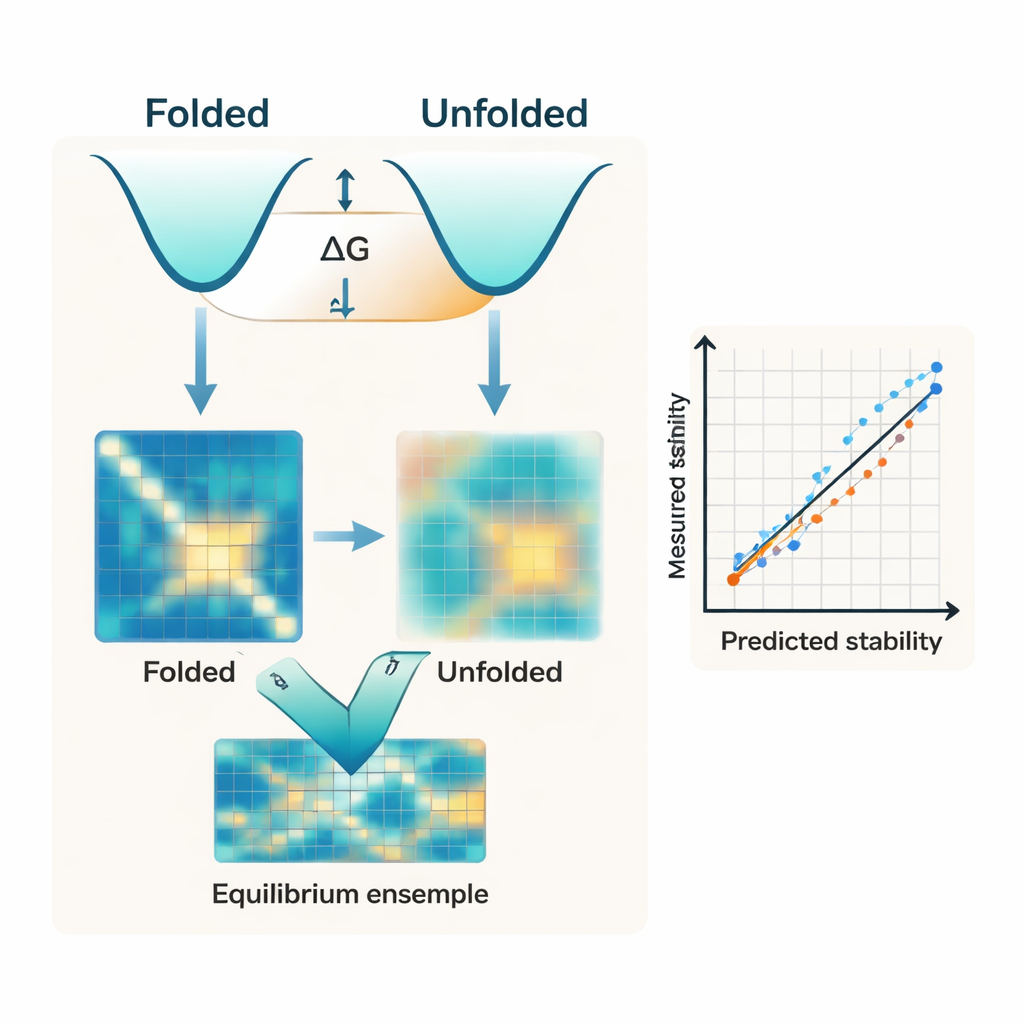
Most modern algorithms focus almost entirely on the folded shape of a protein. They often start from an AI‑predicted structure, such as those from AlphaFold, and treat that single structure as the main determinant of stability. But stability is really the energy gap between two broad ensembles: the compact folded state and the many floppy shapes that make up the unfolded state. The authors argue that ignoring the unfolded side of this balance is a key reason why existing tools struggle to match experimental measurements of folding free energy, known as ΔG.
A new model that learns both states
The researchers introduce IFUM, a deep‑learning system designed to estimate ΔG while also learning what the balance of folded and unfolded states looks like for each protein. Instead of treating the unfolded state as a vague background, IFUM uses ideas from polymer physics to represent it as a “random coil” and encodes both folded and unfolded states as maps of distances between pairs of amino acids. The model takes in information from powerful pre‑trained sequence and structure networks, then jointly predicts the total stability and a probability map describing how much of the protein population is folded versus unfolded at each residue pair. Training on a very large dataset of small, experimentally characterized proteins and known disordered proteins helps IFUM recognize both well‑structured and floppy sequences.
Better numbers and broader mutation coverage
When tested on a carefully controlled dataset of small proteins, IFUM predicts experimental ΔG values with lower error and higher correlation than previous AI‑based methods that rely only on the folded structure or on language models trained on sequences. Crucially, the model also handles a wide variety of sequence changes. It accurately captures the effects of single and double point mutations, as well as insertions and deletions that change protein length—situations where many existing tools either fail outright or were never designed to operate. An internal comparison shows that removing the unfolded‑state objective significantly worsens performance, underscoring that explicitly modeling the unfolded ensemble is not just a conceptual nicety but central to the accuracy of the predictions.
From design bench to real‑world tests
To see whether IFUM can guide real protein engineering, the authors apply it to three challenging design problems: stabilizing interferon‑lambda proteins, reshaping the immune signaling protein IL‑10, and improving a sugar‑modifying enzyme called UGT76G1. In all three cases, IFUM’s predicted stabilities track well with measured melting temperatures, which report how much heat a protein can withstand before unfolding. The model also helps screen hundreds of brand‑new, computer‑designed proteins to pick those most likely to fold and remain soluble in cells, outperforming widely used confidence scores from structure‑prediction networks. These results suggest that IFUM can be used as a practical “stability filter” alongside structure‑based checks in modern protein design workflows.
Limits and future directions
Like any model, IFUM has boundaries. It is trained mainly on short, single‑chain, soluble proteins, and its absolute stability numbers become less trustworthy for much larger proteins or those with extensive flexible loops or membrane‑spanning regions. Its description of the unfolded state is still a simplified statistical model rather than a fully realistic picture of all possible shapes. Nonetheless, the approach demonstrates that teaching an AI to consider both folded and unfolded ensembles yields more reliable stability estimates. For non‑experts, the key takeaway is that IFUM moves us closer to being able to ask a computer, with quantitative confidence, “Will this protein design actually hold together?”, potentially speeding the development of safer biologic drugs and more robust industrial enzymes.
Citation: Lee, H., Cho, Y., Yun, J. et al. Protein folding stability estimation with explicit consideration of unfolded states. Nat Commun 17, 1883 (2026). https://doi.org/10.1038/s41467-026-68637-4
Keywords: protein stability, protein folding, deep learning, protein design, mutations