Clear Sky Science · en
Ligand-controlled regiodivergent and enantioselective C–H cyanation of secondary amines
Why changing tiny bonds matters for big medicines
Many best-selling medicines contain small, nitrogen‑based building blocks called amines. Subtle changes in how the atoms are connected around these amines can turn a weak drug into a powerful, precise treatment—or into something inactive or even harmful. This paper describes a new way to modify these amine building blocks at will, letting chemists choose exactly where on the molecule to react and which mirror-image form to make, both of which are crucial for designing safer and more effective drugs.
Picking one spot on a crowded molecule
Amines in medicines often have several very similar carbon–hydrogen (C–H) bonds that normally behave almost the same. Chemists would like to swap just one of those hydrogens for a useful group, such as a “cyano” (–CN) group, without disturbing the rest of the molecule. That’s difficult because standard reactions tend to go to the most reactive site set by the molecule’s structure, not by the chemist’s choice. Here, the authors work with simple, flexible secondary amines that carry two different carbon chains on nitrogen. They show that, starting from the same amine, they can direct the reaction to one of two neighboring positions—either next to a small N‑methyl group (the so‑called α′ site) or one carbon further away on the other chain (the β site)—simply by changing the ligand that surrounds a copper catalyst.
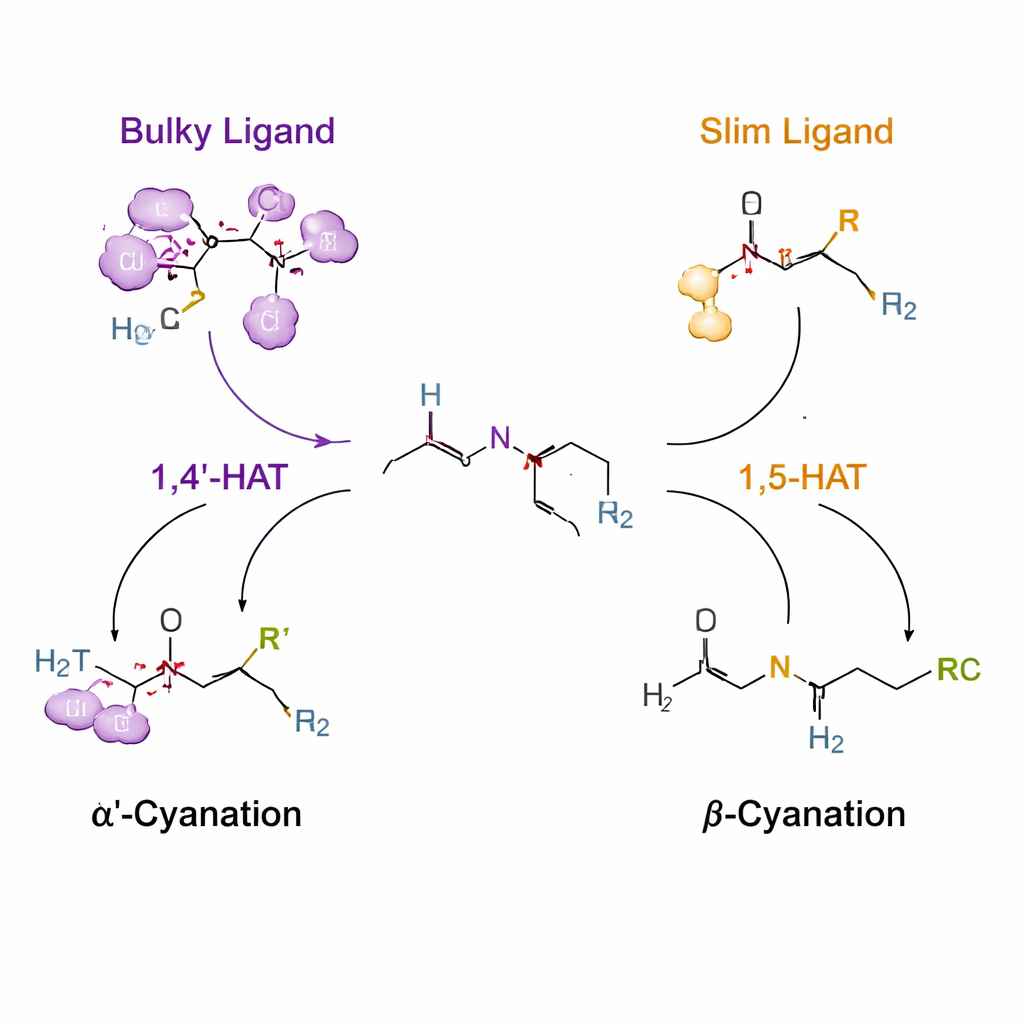
Using controlled hydrogen “hops” to guide reactivity
The key trick relies on a process called hydrogen atom transfer, where a short‑lived nitrogen‑centered radical plucks a hydrogen from a nearby carbon. Normally, such radicals prefer a particular distance, favoring a six‑membered “reach” known as 1,5‑HAT. The authors attach a temporary urea and chlorine handle to the amine so that, under copper catalysis, this nitrogen radical forms and can grab hydrogen either from the α′ or β position. By designing ligands—organic molecules that wrap around the copper—they reshape the environment of the radical. A very bulky ligand (named L14) nudges the system toward an unusual 1,4′‑HAT step that targets the N‑methyl group, giving selective α′‑cyanation. Slimmer ligands (such as L8) allow the conventional 1,5‑HAT path, steering the reaction to the β position instead.
From control of position to control of handedness
Beyond choosing where to react, the team also wants control over handedness, or chirality, which is vital because many drugs exist as left‑ and right‑handed forms that behave differently in the body. To achieve this, they introduce chiral ligands—molecules that themselves have a handed shape—into the copper complex. Two such ligands, L24 and L41, give high preference for one mirror image when the reaction installs a cyano group at β positions, including both benzylic sites (next to aromatic rings) and allylic sites (next to carbon–carbon double bonds). Across a wide variety of amine starting materials, the method delivers β‑cyanated products with excellent selectivity for both site and handedness, and it works on gram scale, showing that the process is practical and robust for synthesis.
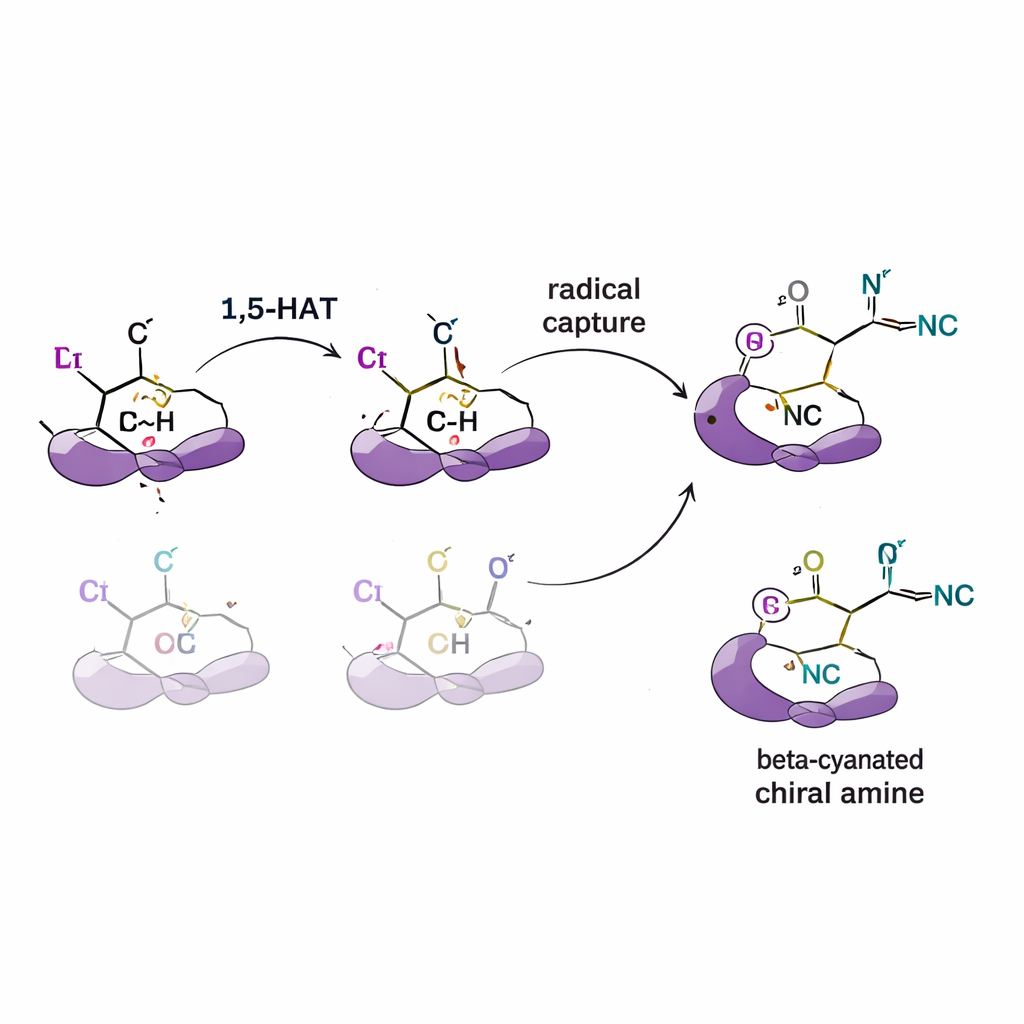
Testing the mechanism behind the selectivity
To figure out how this control arises, the authors carry out a series of mechanistic experiments. By adding radical “traps,” they confirm that reactive radical intermediates are indeed involved. Using substrates in which certain hydrogens are replaced with deuterium (a heavier form of hydrogen), they detect kinetic isotope effects that point to the hydrogen transfer step as the slow, selectivity‑setting part of the reaction. Labeling experiments also show that hydrogen moves in a single, one‑way step rather than shuttling back and forth between positions. Complementary computer simulations (density functional theory) support these findings, indicating that the shape and bulk of the ligand alter the energy of competing hydrogen‑transfer pathways and the way the radical then combines with copper and cyanide to give a preferred mirror image.
What this means for future drug design
Overall, this work introduces a flexible strategy to re‑engineer common amine groups at two closely related sites, on demand, with fine control over molecular handedness. By swapping only the ligand on a copper catalyst, chemists can choose whether to place a cyano group on a small N‑methyl unit or on the neighboring carbon of a different side chain, and can do so across many complex, drug‑like molecules. Because cyano groups are valuable stepping stones to many other functional groups, this “dial‑a‑site” and “dial‑a‑hand” approach should make it easier to explore and optimize new medicines built from the same basic amine skeletons.
Citation: Mao, YJ., Chen, X., Li, HL. et al. Ligand-controlled regiodivergent and enantioselective C–H cyanation of secondary amines. Nat Commun 17, 1869 (2026). https://doi.org/10.1038/s41467-026-68598-8
Keywords: amine functionalization, hydrogen atom transfer, copper catalysis, enantioselective cyanation, medicinal chemistry