Clear Sky Science · en
Genomic language model mitigates chimera artifacts in nanopore direct RNA sequencing
Why cleaning up RNA reads matters
Our cells constantly read genetic instructions written in RNA, and new sequencing technologies now let scientists watch this process in unprecedented detail. One of the most powerful tools, nanopore direct RNA sequencing, can read whole RNA molecules in one go—but it also introduces glitches that can make it look like genes are broken and re-stitched in ways that never happen in real life. This study introduces DeepChopper, a software tool that acts like a language model for genomes, cleaning up these errors so researchers can trust what they see in RNA data.
When the sequencer invents fake gene mashups
Modern nanopore machines pull individual RNA strands through tiny pores and read out their sequence directly. This has big advantages over older methods, such as preserving chemical modifications and capturing full-length transcripts in a single read. But the process also relies on short helper pieces called adapters that are glued to RNA molecules during library preparation. Sometimes, two or more RNA molecules get accidentally joined together by these adapters, creating what look like chimera—hybrid molecules that appear to fuse different genes. Standard analysis tools can misinterpret these technical leftovers as real biological events, like cancer-related gene fusions or unusual splicing patterns, leading to misleading results.
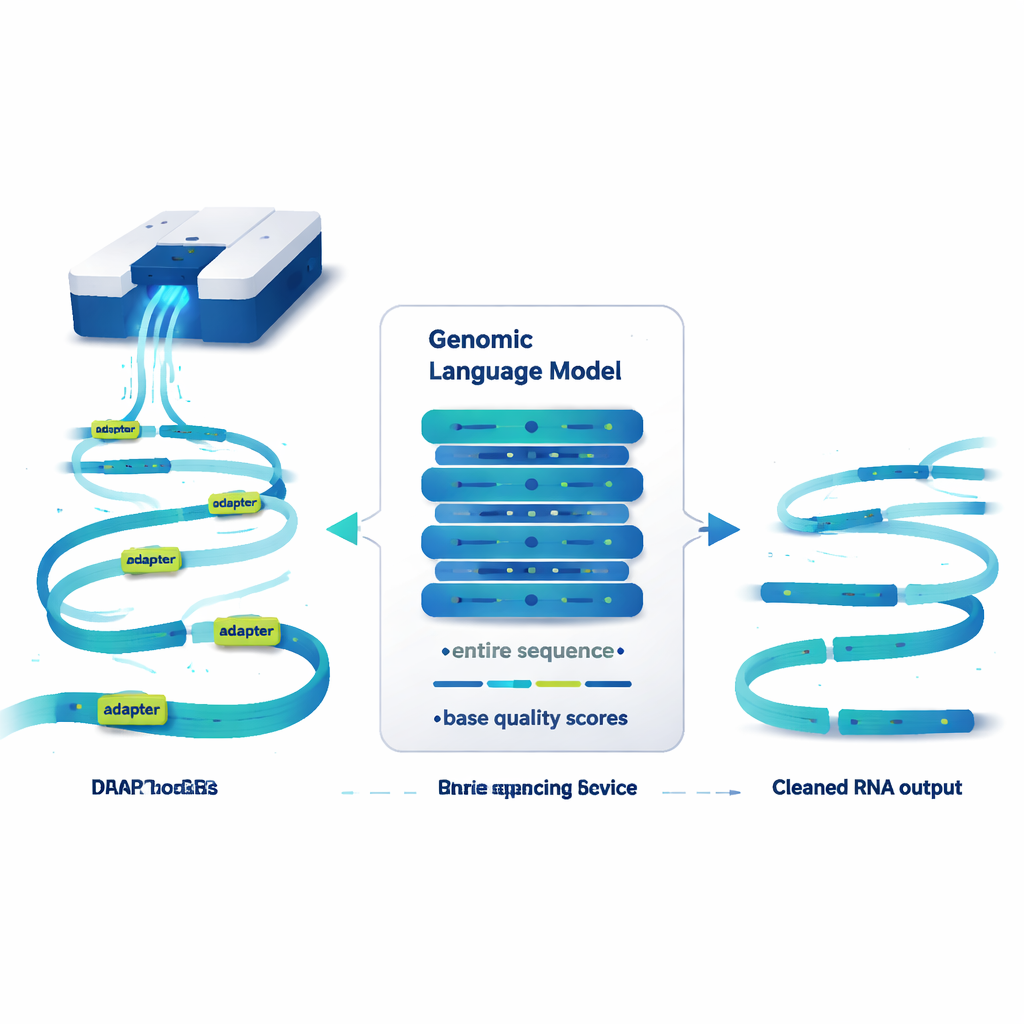
A language model that reads genomes, not sentences
DeepChopper treats genetic sequences a bit like text and applies ideas from large language models to them. Instead of working with words, it reads RNA sequences one letter at a time, along with a quality score for each letter that indicates how reliable the read is. Built on a compact architecture called HyenaDNA, it can scan up to 32,000 bases at once—long enough to cover essentially any human RNA molecule. For every single position, DeepChopper estimates whether that base is part of a genuine RNA sequence or part of an adapter. A refinement step then smooths these predictions so that adapters are marked as continuous blocks rather than scattered spots.
Cutting out the bad joins without throwing away data
Once DeepChopper has found adapters inside a read, it does something crucial: instead of discarding the whole read, it “chops” at those adapter sites and keeps the real pieces. This way, an artificial fusion of two RNAs can be split back into its original parts. In tests on millions of nanopore reads from multiple human cancer cell lines and stem cells, DeepChopper far outperformed existing adapter-trimming tools, which were never designed for this direct RNA setting. It correctly recognized adapters with over 99% precision and recall on synthetic benchmarks, and it scaled efficiently to datasets with more than 20 million reads using graphics processors.
Separating real gene fusions from sequencing mirages
The authors then asked whether DeepChopper could distinguish genuine biological events from artifacts in real cancer data. By comparing direct RNA reads with matching datasets produced by independent methods (such as direct cDNA sequencing on both Oxford Nanopore and PacBio platforms), they could label which apparent chimeras were supported by other technologies and which were not. DeepChopper reduced unsupported chimeric alignments by as much as 62–91%, while greatly enriching the fraction that were confirmed by other methods. It also slashed the number of suspicious gene fusion calls by nearly 90%, especially those involving ribosomal genes that turned out to be frequent artifacts. At the same time, true fusion events backed by short-read RNA sequencing were preserved.
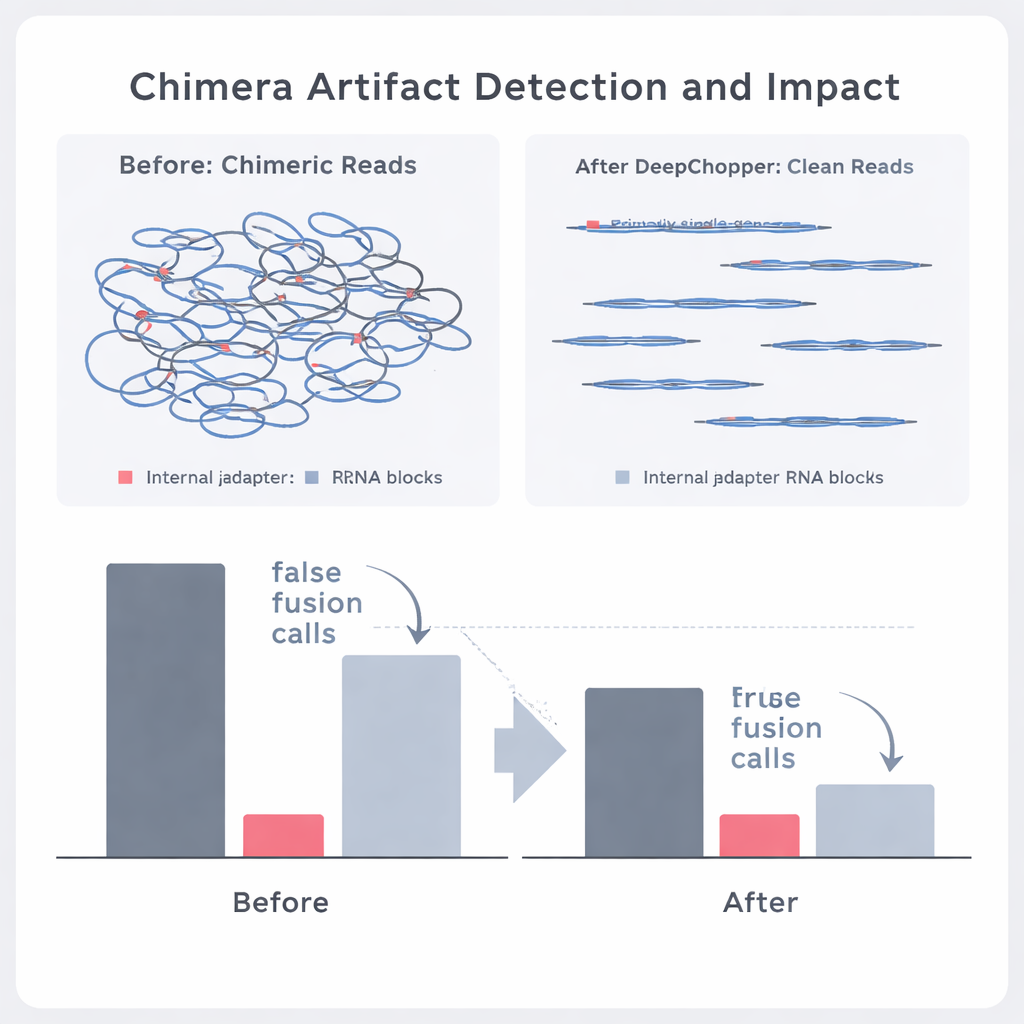
Better chemistry helps—but artifacts remain
Oxford Nanopore recently released an updated sequencing kit (RNA004) designed in part to reduce technical artifacts. DeepChopper was first applied “out of the box” to data from this new chemistry and still found that a small but important fraction of reads contained internal adapters and chimeric joins. Even without extra training, the model cut artifactual chimeras by about one fifth; after fine-tuning on the new data, it performed slightly better, all while leaving genuine signals intact. Across all chemistries and cell types, correcting these artifacts allowed downstream tools to detect many more full-length and alternative transcripts, giving a clearer view of the cell’s RNA landscape.
What this means for future RNA studies
For non-specialists, the key message is that not every surprising RNA connection reported by a sequencer is real biology—some are wiring errors introduced by the technology itself. DeepChopper acts like a highly trained copy editor for nanopore RNA data, spotting the telltale adapter sequences that join unrelated molecules and cutting them out with single-base precision. The result is cleaner, more trustworthy maps of which RNA molecules exist in a cell and how they are put together. As labs increasingly rely on long-read RNA sequencing to study cancer, brain disorders, and other complex diseases, tools like DeepChopper will be essential for turning noisy raw reads into reliable biological insight.
Citation: Li, Y., Wang, TY., Guo, Q. et al. Genomic language model mitigates chimera artifacts in nanopore direct RNA sequencing. Nat Commun 17, 1864 (2026). https://doi.org/10.1038/s41467-026-68571-5
Keywords: nanopore RNA sequencing, chimeric reads, gene fusion artifacts, genomic language model, DeepChopper