Clear Sky Science · en
Endothelial FUNDC1 regulates metabolic reprogramming and the obesity-diabetes transition through the SIRT3/GATA2/endothelin-1 axis
Why blood vessel cells matter for obesity and diabetes
Obesity and type 2 diabetes are often blamed on fat tissue, the liver, or the pancreas. But this study reveals a surprising culprit: the thin layer of cells lining our blood vessels, called endothelial cells. The researchers show that a small mitochondrial protein in these cells, named FUNDC1, can help push the body from simple weight gain into full-blown diabetes by changing how blood vessels talk to metabolic organs.
Stress on the body’s inner “skin”
Endothelial cells form a vast, delicate network that controls blood flow, nutrient delivery, and signals to nearby tissues. Under healthy conditions, they balance relaxing factors like nitric oxide with tightening factors like endothelin-1 (ET-1). In obesity and early diabetes, this balance tips toward ET-1, which not only narrows blood vessels but also disrupts how fat, muscle, and liver cells handle sugar and fat. The authors began by showing in mice that blood vessel dysfunction appears after just two months on a high-fat diet, before clear insulin resistance develops, suggesting that injured endothelium may help trigger metabolic disease rather than simply respond to it.
A mitochondrial switch that shapes body fat
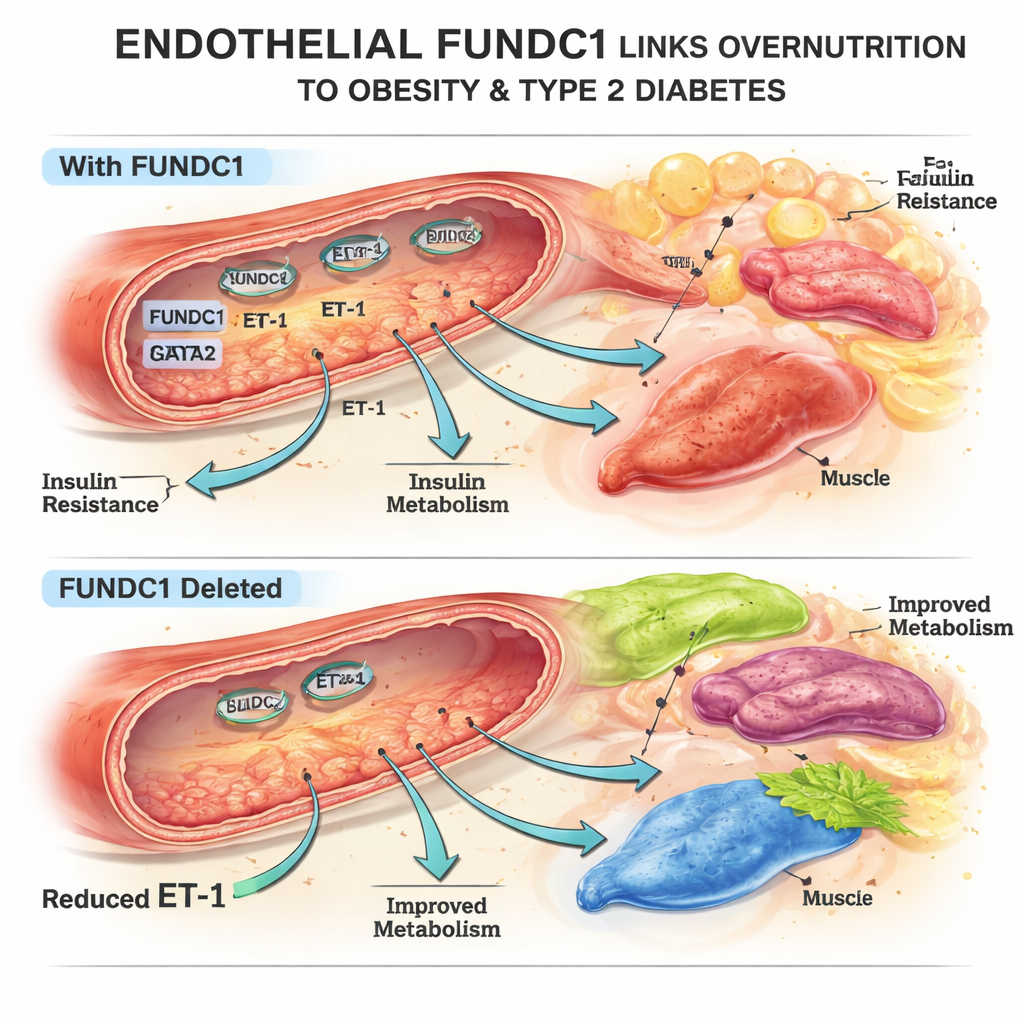
The team focused on FUNDC1, a protein on the surface of mitochondria, the cell’s energy factories. In mouse and human endothelial cells exposed to excess fat, FUNDC1 levels changed over time: they dropped at first, then rose strongly with prolonged overnutrition. Using genetically engineered mice that lack FUNDC1 only in endothelial cells, the researchers discovered that these animals were partly protected from high-fat diet weight gain, had less body fat, smaller fat cells, and better blood sugar control. Their fat, liver, and brown fat tissues responded more strongly to insulin, even though insulin levels themselves were not higher. These changes could not be explained by differences in food intake or activity, pointing instead to how the vasculature was influencing metabolism.
A chemical messenger that drives insulin resistance
To find out how endothelial FUNDC1 affects distant organs, the authors screened several substances secreted by endothelial cells. One stood out: ET-1. When FUNDC1 was deleted in endothelial cells, ET-1 production in blood vessels and in the bloodstream fell markedly, both in normal and high-fat diet conditions. Experiments in cultured fat cells, liver cells, and muscle cells showed that ET-1 promoted pre-fat-cell growth, altered fat storage and breakdown, and worsened fat buildup in liver and muscle exposed to high fat—a pattern known to foster insulin resistance. In live mice, infusing ET-1 at the start of a high-fat diet erased the protective benefits of endothelial FUNDC1 deletion: body weight, fat mass, blood sugar control, and vessel function all deteriorated, underscoring ET-1 as a key link between the endothelium and metabolic disease.
An internal signaling axis: FUNDC1, SIRT3, and GATA2
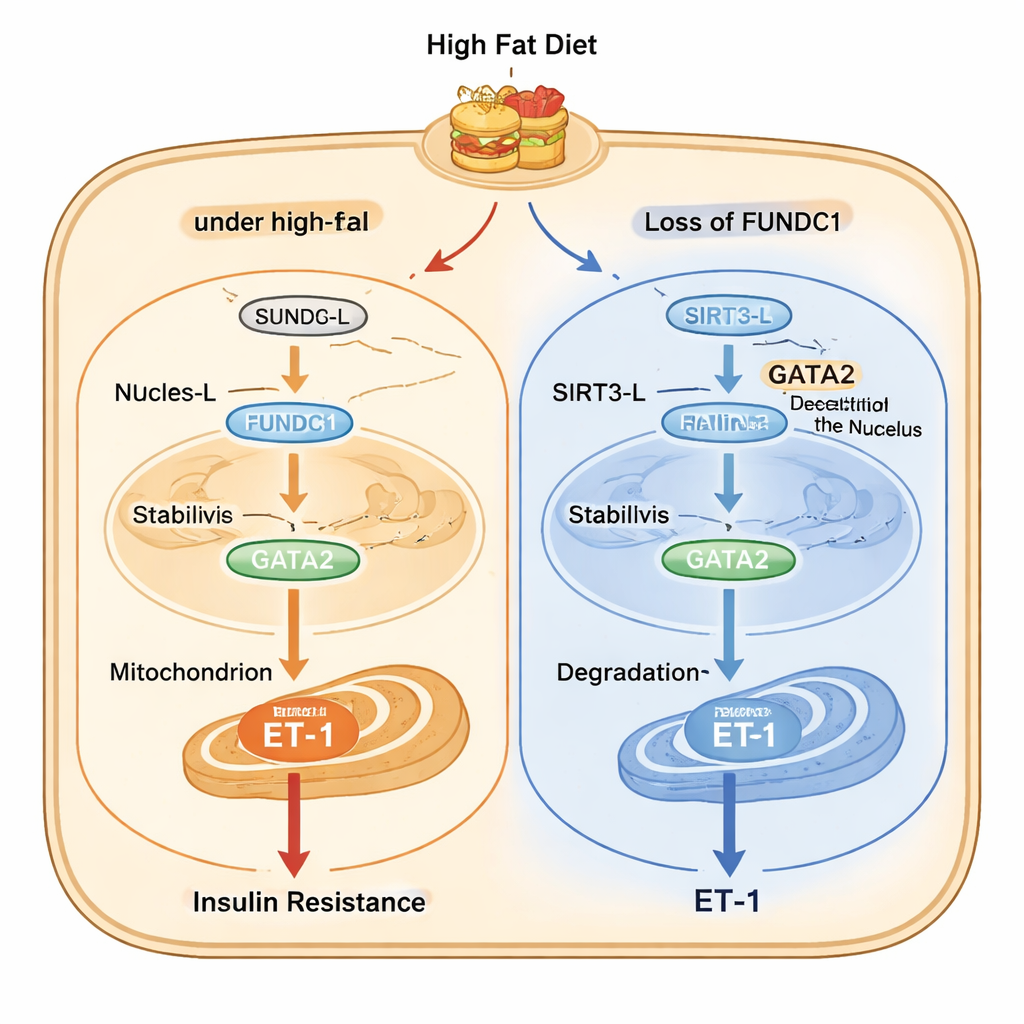
The study then maps out a detailed molecular chain inside endothelial cells. Under high-fat stress, a long form of the enzyme SIRT3 (SIRT3-L), which can reside in both the nucleus and mitochondria, moves from the nucleus into mitochondria with the help of FUNDC1 and a chaperone protein called HSC70. Once sequestered in mitochondria, less SIRT3-L is available in the nucleus to remove acetyl groups from GATA2, a transcription factor that boosts ET-1 gene activity. More acetylated GATA2 is more stable and drives more ET-1 production. When FUNDC1 is absent, SIRT3-L stays in the nucleus, where it deacetylates GATA2, leading to GATA2 breakdown and lower ET-1 output. Intriguingly, SIRT3 in turn promotes FUNDC1 degradation, creating a feedback loop that normally restrains the pathway but becomes dysregulated during chronic overnutrition.
From mouse models to human disease
To test whether this mechanism matters in people, the researchers examined blood and small arteries from individuals with obesity and type 2 diabetes and from healthy volunteers. Patients with both conditions had higher blood levels of ET-1 and higher expression of FUNDC1, GATA2, and the ET-1 gene in their vessel lining. The amount of ET-1 in blood tracked closely with body mass index and long-term blood sugar (HbA1c), and vessel ET-1 gene levels correlated strongly with FUNDC1 and GATA2. These patterns mirror the mouse findings and support the idea that an overactive FUNDC1–SIRT3–GATA2–ET-1 axis is at work in human vascular tissue under metabolic stress.
A new target in the fight against diabetes
For non-specialists, the core message is that damage from overeating may first show up in the cells lining our blood vessels. There, a mitochondrial protein, FUNDC1, helps redirect a regulatory enzyme, SIRT3, away from the nucleus, allowing another factor, GATA2, to turn up production of ET-1, a powerful hormone-like signal that promotes both vessel stiffening and insulin resistance. Blocking this pathway—by reducing endothelial FUNDC1 activity or dialing down ET-1—could help prevent the transition from obesity to diabetes and protect blood vessels at the same time.
Citation: Li, J., Li, D., Zhao, F. et al. Endothelial FUNDC1 regulates metabolic reprogramming and the obesity-diabetes transition through the SIRT3/GATA2/endothelin-1 axis. Nat Commun 17, 1836 (2026). https://doi.org/10.1038/s41467-026-68548-4
Keywords: endothelial cells, mitochondria, endothelin-1, obesity, type 2 diabetes