Clear Sky Science · en
Palmitic acid-triggered B7H3 palmitoylation promotes immune escape
Why this research matters for cancer patients
Most people have heard that new immunotherapy drugs can help the immune system attack cancer, yet many patients with common colorectal (bowel) cancer see little benefit. This study uncovers a hidden biochemical trick that colorectal tumors use to shield themselves from immune attack, and it points to a new type of drug—a small peptide—that could make immunotherapy work better for those patients.
A roadblock in current bowel cancer immunotherapy
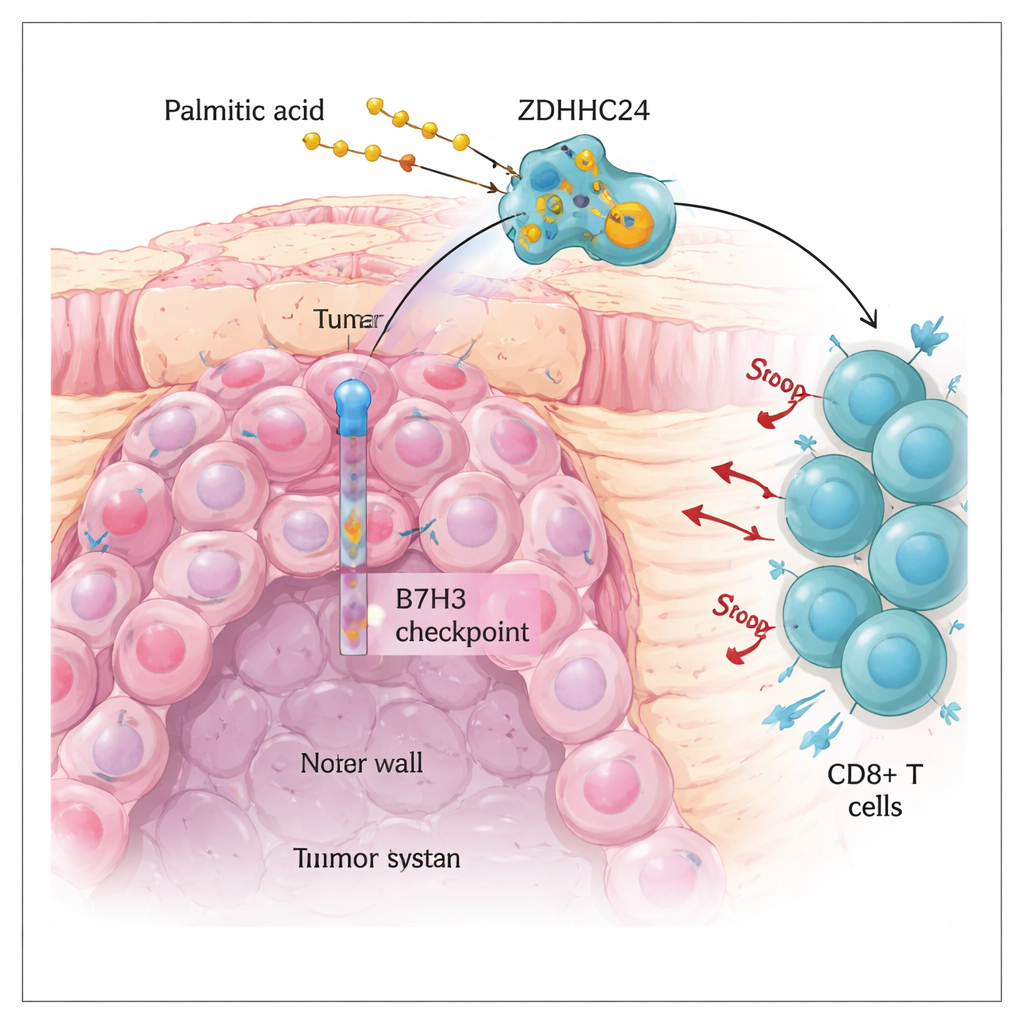
Colorectal cancer is one of the most common cancers worldwide. Immunotherapy drugs that release immune “brakes,” such as PD-1 and PD-L1 inhibitors, have transformed care for a minority of patients whose tumors carry many DNA mistakes (so‑called MSI-H tumors). Unfortunately, most colorectal cancers are microsatellite-stable (MSS) and respond poorly to these treatments. The authors focused on another brake molecule called B7H3, which sits on the surface of tumor cells and dampens the activity of killer immune cells, especially CD8+ T cells. They found that B7H3 protein is strongly elevated in MSS tumors and linked to worse survival, even though the underlying B7H3 gene is not more active. That mismatch pointed to an important question: what keeps the B7H3 protein so abundant on tumor cells?
How a common fat helps tumors hide
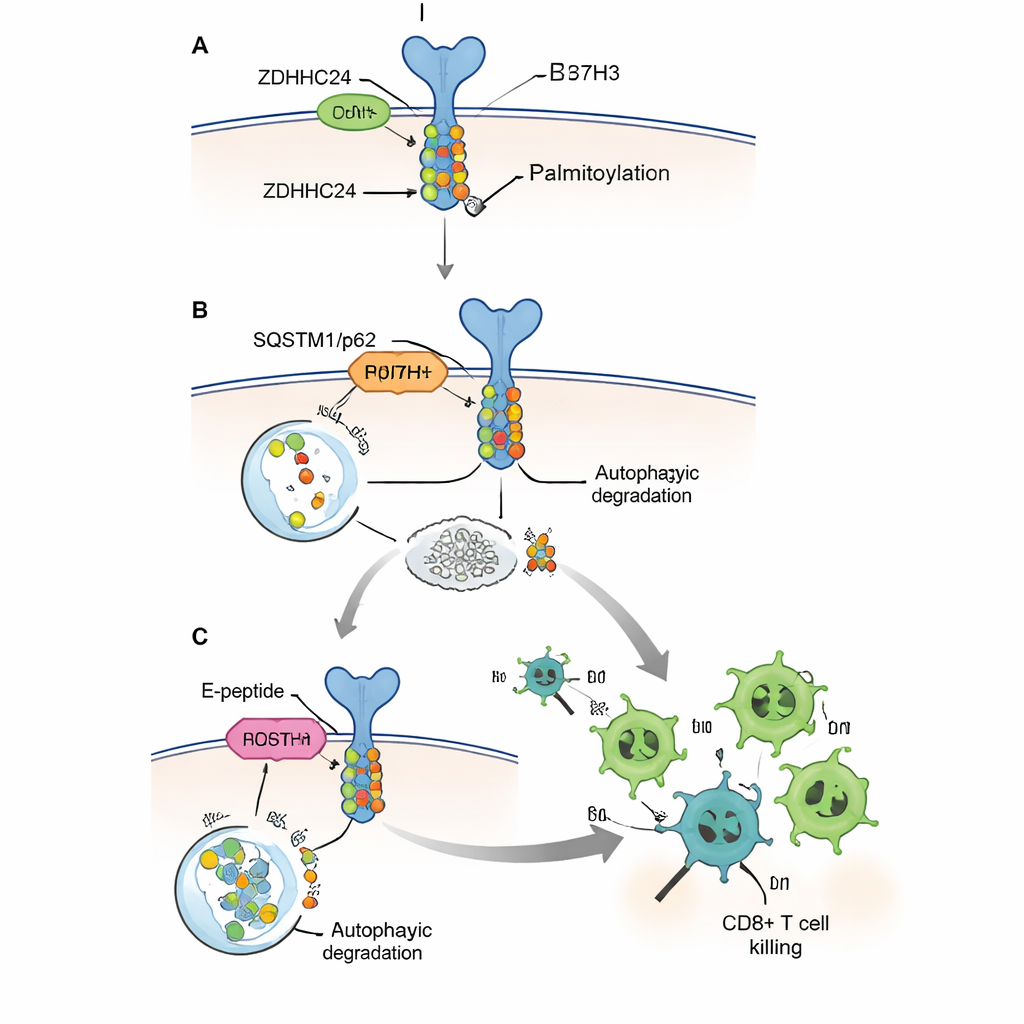
The team suspected that tumor metabolism might be stabilizing B7H3. By comparing the gene activity and small-molecule profiles of MSS and MSI-H colorectal tumors, they discovered that many fat‑related pathways differ, with one fatty acid, palmitic acid, standing out. When they exposed colon cancer cells to several fatty breakdown products in the lab, only palmitic acid caused a clear rise in B7H3 protein levels. Further experiments showed why: palmitic acid feeds into a chemical modification process called palmitoylation, in which a fat chain is attached to specific spots on proteins. This modification, carried out by an enzyme named ZDHHC24 at a single cysteine position within B7H3, made B7H3 more stable and more abundant on the tumor-cell surface.
Blocking the trash route for an immune brake
Cells normally remove unwanted or damaged proteins through systems that function like recycling and garbage disposal. The researchers showed that B7H3 is mainly broken down by a cellular “self‑eating” pathway called autophagy, which uses a receptor protein called SQSTM1/p62 to tag cargo for destruction. When B7H3 was palmitoylated, it bound poorly to this receptor and escaped autophagic degradation, leading to persistent, high levels of the immune brake. When the palmitoylation site was mutated so it could no longer carry the fat tag, or when the ZDHHC24 enzyme was deleted, B7H3 was more efficiently routed to cell “trash bags” and broken down. In mice, tumors lacking palmitoylated B7H3 grew more slowly, did not change their intrinsic growth rate in immune‑deficient animals, and showed dramatically more CD8+ T cells and stronger tumor‑killing activity. This demonstrated that the modification mainly acts by disarming immunity, not by altering how fast the cancer cells divide.
Turning down the shield and boosting T cells
Because fully eliminating ZDHHC24 or B7H3 with genetics is not a practical treatment in patients, the authors designed a short, cell‑penetrating peptide they call the E‑peptide. It mimics a small stretch of the B7H3 protein that normally binds to ZDHHC24, acting as a decoy that prevents the enzyme from modifying real B7H3 molecules. In cultured cells, E‑peptide reduced B7H3 palmitoylation and protein levels, restored its recognition by the autophagy machinery, and allowed human CD8+ T cells to kill tumor cells more efficiently. In several mouse models, including a humanized mouse carrying human immune cells and human colorectal tumors, injections of E‑peptide shrank tumors, increased the number of CD8+ T cells inside them, and boosted the production of key killer molecules such as granzyme B and interferon‑gamma.
A new partner for existing immunotherapy
Finally, the researchers asked whether this strategy could work alongside standard PD‑1 blockade. In mouse models of colorectal cancer, both E‑peptide alone and anti‑PD‑1 antibody alone offered some tumor control. But when combined, the two treatments produced much stronger and longer‑lasting tumor suppression, in some cases causing tumors to disappear and more than doubling average survival compared to controls. This suggests that disabling the B7H3 shield by cutting its fat “anchor” can complement existing checkpoint drugs that target the PD‑1 pathway.
What this means for future cancer treatment
In plain terms, this study shows that a common dietary fat, palmitic acid, can be hijacked by colorectal tumors to chemically armor an immune‑blocking protein (B7H3) and help the cancer hide. By blocking that single chemical attachment, the authors were able to strip away the armor, let the cell’s own disposal systems remove B7H3, and reopen access for CD8+ T cells to attack. While the E‑peptide itself is an early‑stage experimental tool, the work identifies B7H3 palmitoylation—and its enzyme ZDHHC24—as promising drug targets. If similar agents prove safe and effective in humans, they might one day turn more colorectal cancers from “cold” tumors that ignore immunotherapy into “hot” tumors that the immune system can recognize and destroy.
Citation: Rao, Z., Huang, C., Wu, Q. et al. Palmitic acid-triggered B7H3 palmitoylation promotes immune escape. Nat Commun 17, 1810 (2026). https://doi.org/10.1038/s41467-026-68525-x
Keywords: colorectal cancer immunotherapy, B7H3 checkpoint, palmitic acid metabolism, protein palmitoylation, CD8 T cell tumor immunity