Clear Sky Science · en
TET1 as a master regulator controlling GPX4-dependent and -independent ferroptosis surveillance in acute myeloid leukemia
Why this research matters for cancer treatment
Many new cancer drugs aim to push malignant cells into a self-destruct mode called ferroptosis, a type of cell death driven by iron and fat damage. Yet some tumors stubbornly resist this approach. This study reveals how a DNA-modifying protein called TET1 helps leukemia cells evade ferroptosis through two separate biochemical defense systems—and shows that blocking these defenses can make even resistant cancers vulnerable.
A deadly mix of iron and damaged fats
Ferroptosis occurs when iron fuels the uncontrolled oxidation of fats in cell membranes, ultimately causing cells to rupture. In acute myeloid leukemia (AML), as in many cancers, cells deploy powerful surveillance systems to keep this process in check. One key guardian is the enzyme GPX4, which uses a small molecule called glutathione to neutralize harmful lipid peroxides. Other backup systems generate antioxidant molecules that can trap dangerous radicals even when GPX4 is compromised. Understanding which master switches coordinate these defenses is crucial for designing therapies that reliably trigger ferroptosis in cancer cells while sparing healthy tissue.
TET1 emerges as a central control hub
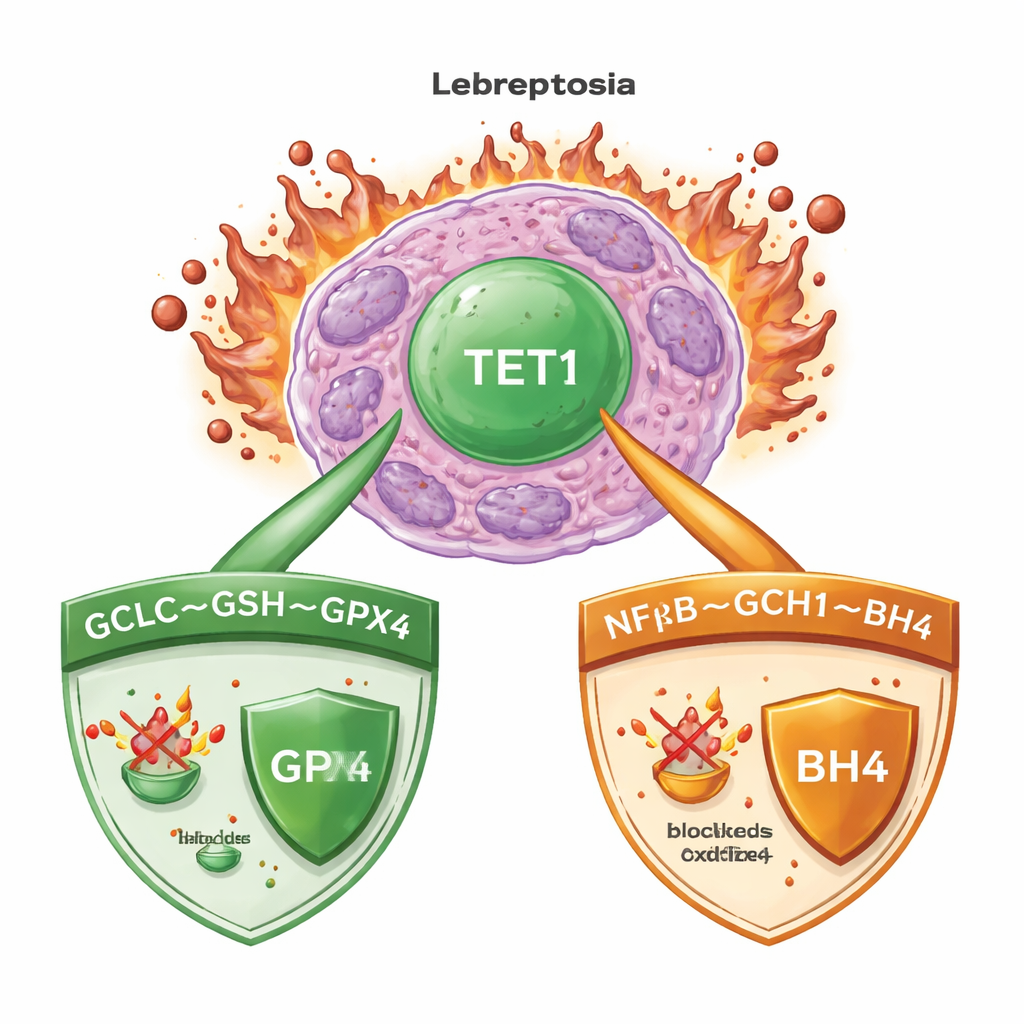
The researchers compared dozens of cancer cell samples, including many AML lines and patient-derived cells, and noticed a clear pattern: cells that resisted ferroptosis had higher levels of TET1, an enzyme that alters DNA chemical marks and influences gene activity. When they reduced TET1 levels using genetic tools or inhibited its activity with a small molecule, cancer cells became markedly more sensitive to ferroptosis-inducing drugs. This held true in both lab dishes and mouse models of AML. Conversely, boosting TET1 expression protected cells from ferroptotic death and limited the buildup of reactive oxygen species, the chemically aggressive by-products that drive membrane damage.
Strengthening the main antioxidant shield
Digging deeper, the team mapped where TET1 acts on the genome and found that it directly activates a gene called GCLC. GCLC encodes a critical enzyme that launches production of glutathione, the fuel for GPX4. By increasing a specific DNA mark (5-hydroxymethylcytosine) at the GCLC promoter, TET1 ramps up glutathione synthesis. Under normal nutrient conditions, this boosts the cell’s main antioxidant pool. Under cystine starvation, the same enzyme complex also makes unusual γ-glutamyl–peptides that help mop up excess glutamate, another way to blunt ferroptosis. In both cultured cells and mice, loss of TET1 or pharmacologic inhibition of glutathione synthesis sharply lowered glutathione levels and these protective peptides, making leukemia cells far more vulnerable to ferroptosis triggers.
A second, GPX4-independent escape route
Surprisingly, TET1’s protective role did not end with the glutathione–GPX4 axis. Even when GPX4 itself was removed from leukemia cells, extra TET1 could still prevent ferroptotic death, hinting at a second line of defense. The authors traced this back to TET1’s activation of the NFκB signaling pathway, in particular a component called NFKB2. This, in turn, boosts expression of GCH1, an enzyme that produces the antioxidant molecule BH4. BH4 can protect membrane lipids from oxidation without relying on GPX4. When GCH1 was genetically silenced or chemically blocked, the ability of TET1 to shield cells from ferroptosis was partially lost. Together, these findings define a TET1–NFKB2–GCH1 route that forms a GPX4-independent ferroptosis surveillance system.
Turning a weakness into a therapeutic opportunity
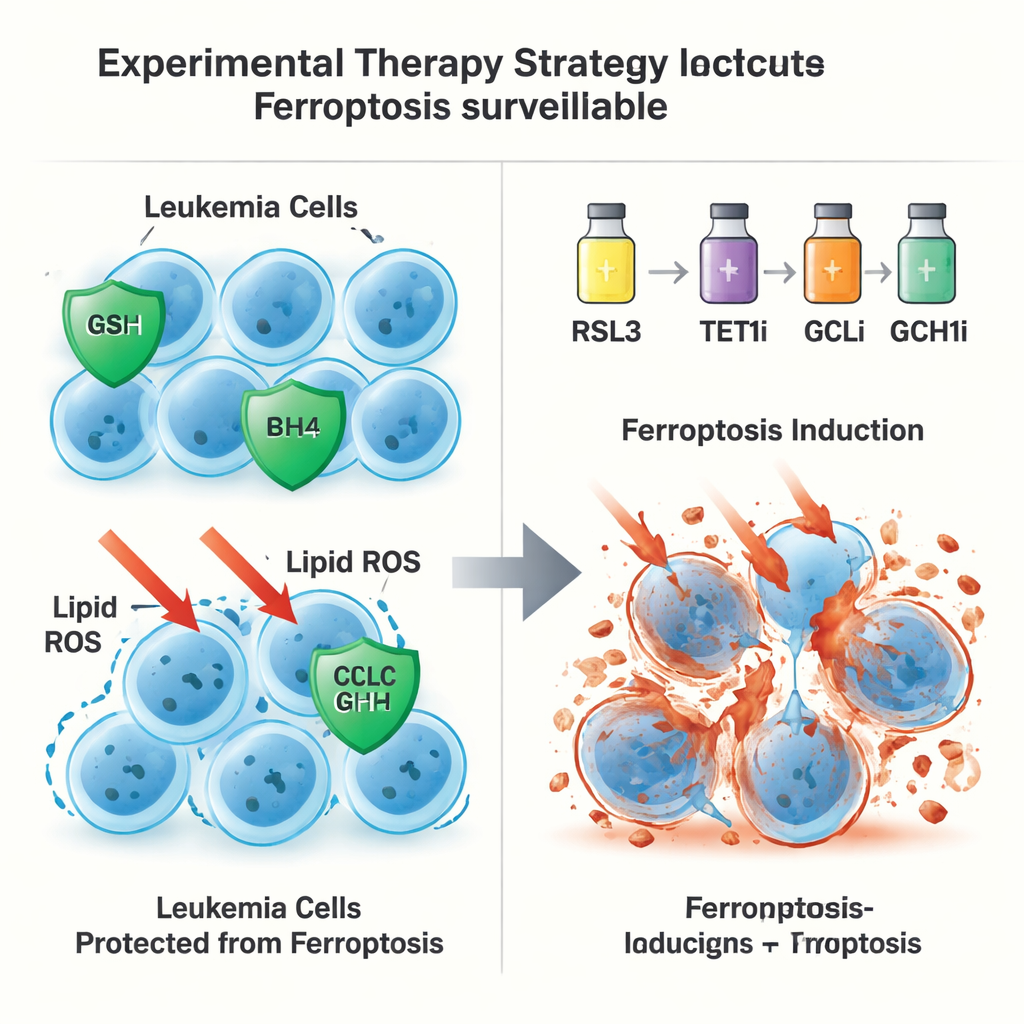
Armed with this dual-pathway map, the researchers tested whether simultaneously nudging ferroptosis and weakening TET1-controlled defenses could provide a therapeutic edge. In mouse AML models and patient-derived leukemia grafts in mice, low doses of a ferroptosis-inducing drug combined with inhibitors of TET1, GSH synthesis (via GCLC), or GCH1 dramatically reduced leukemia burden, prolonged survival, and depleted leukemia-initiating cell populations. Importantly, the ferroptosis inducer was used at a fraction of doses reported in prior studies, reducing concerns over toxicity to normal blood stem cells.
What this means for future cancer therapies
For non-specialists, the key message is that leukemia cells survive by running two overlapping antioxidant “shield” systems, both coordinated by TET1: one centered on glutathione and GPX4, and another based on GCH1 and BH4. This work shows that by modestly activating ferroptosis while at the same time blocking TET1 and its downstream partners, doctors may one day be able to overcome resistance and selectively push cancer cells over the edge, without overwhelming healthy tissues. Although these strategies are not yet ready for the clinic, the study identifies TET1 as a powerful control node and a promising target for combination therapies across AML and potentially other hard-to-treat cancers.
Citation: Yang, L., Lu, J., Yun, W. et al. TET1 as a master regulator controlling GPX4-dependent and -independent ferroptosis surveillance in acute myeloid leukemia. Nat Commun 17, 1800 (2026). https://doi.org/10.1038/s41467-026-68509-x
Keywords: ferroptosis, acute myeloid leukemia, TET1, glutathione, cancer epigenetics