Targeted drugs have transformed treatment for certain lung cancers by homing in on a faulty growth signal called EGFR. Yet for most patients, these drugs stop working within a couple of years as the cancer evolves resistance. This study uncovers a surprising twist: once tumors become resistant to EGFR inhibitors, they develop a new Achilles’ heel that can be attacked with a different type of compound. Understanding this hidden weakness could inspire future treatment strategies that corner cancer evolution instead of always chasing it.
A hidden weakness revealed
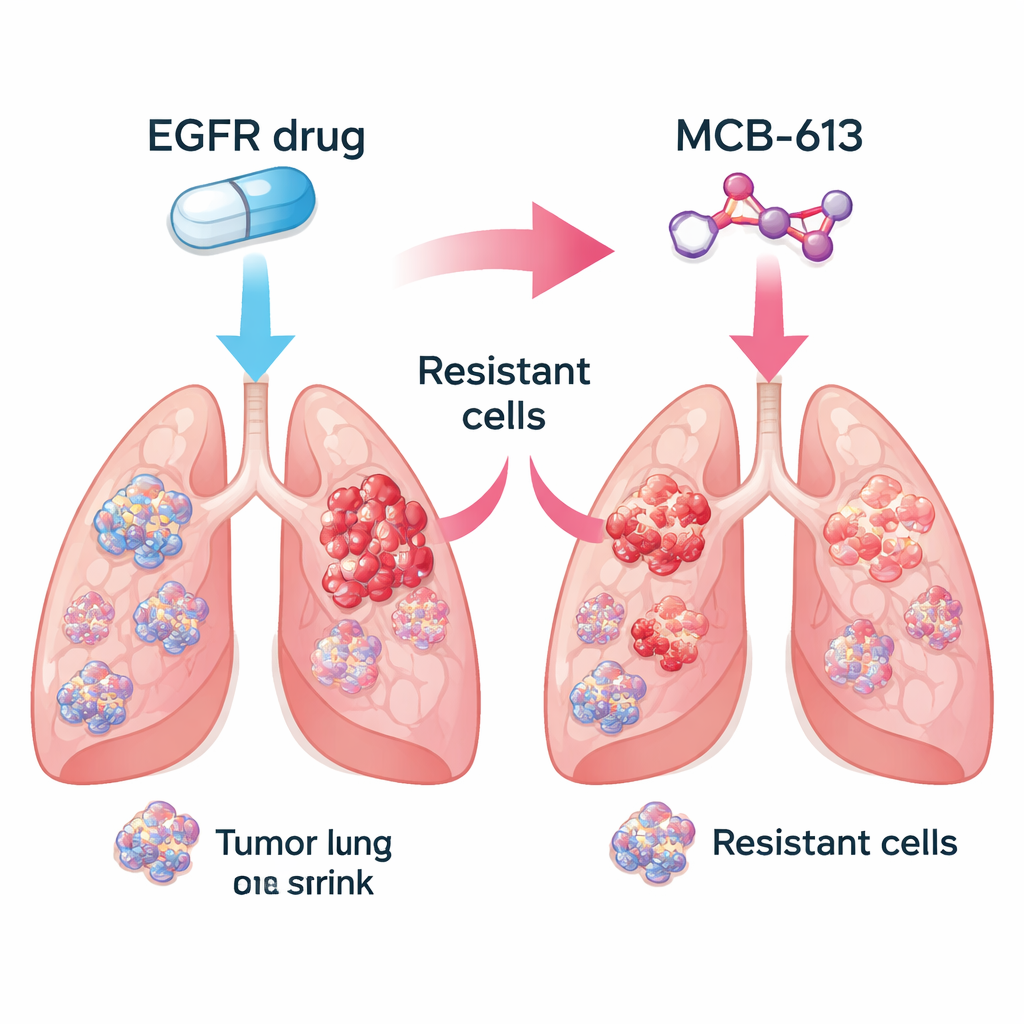
The researchers focused on non-small cell lung cancers driven by mutant EGFR, a common form of the disease. In the lab, they compared drug-sensitive cancer cells with closely related cells that had evolved resistance to EGFR-blocking medicines such as gefitinib and osimertinib. They then tested a library of about 2,100 small molecules to see which ones killed the resistant cells more effectively than the original, drug-sensitive cells. Among many candidates, one compound, called MCB-613, consistently stood out. Resistant cells that shrugged off EGFR inhibitors turned out to be unusually vulnerable to MCB-613, both in lab dishes and in mouse tumors.
Trapping mixed tumor populations Figure 1.
Real tumors are mixtures of cells: some remain sensitive to the original drug, while others acquire resistance through different genetic tricks. The team asked whether combining an EGFR inhibitor with MCB-613 could wipe out this diversity. In a controlled experiment, they mixed mostly drug-sensitive cells with a small fraction of multiple resistant types, mimicking a patient’s tumor. Treating this mixed population with either the EGFR inhibitor or MCB-613 alone allowed some cells to survive and grow. But when both agents were used together, the entire population collapsed. This suggests that pairing a standard targeted therapy with a carefully chosen “collateral sensitivity” drug might push tumors into an evolutionary dead end.
A molecular bridge that breaks a guardian
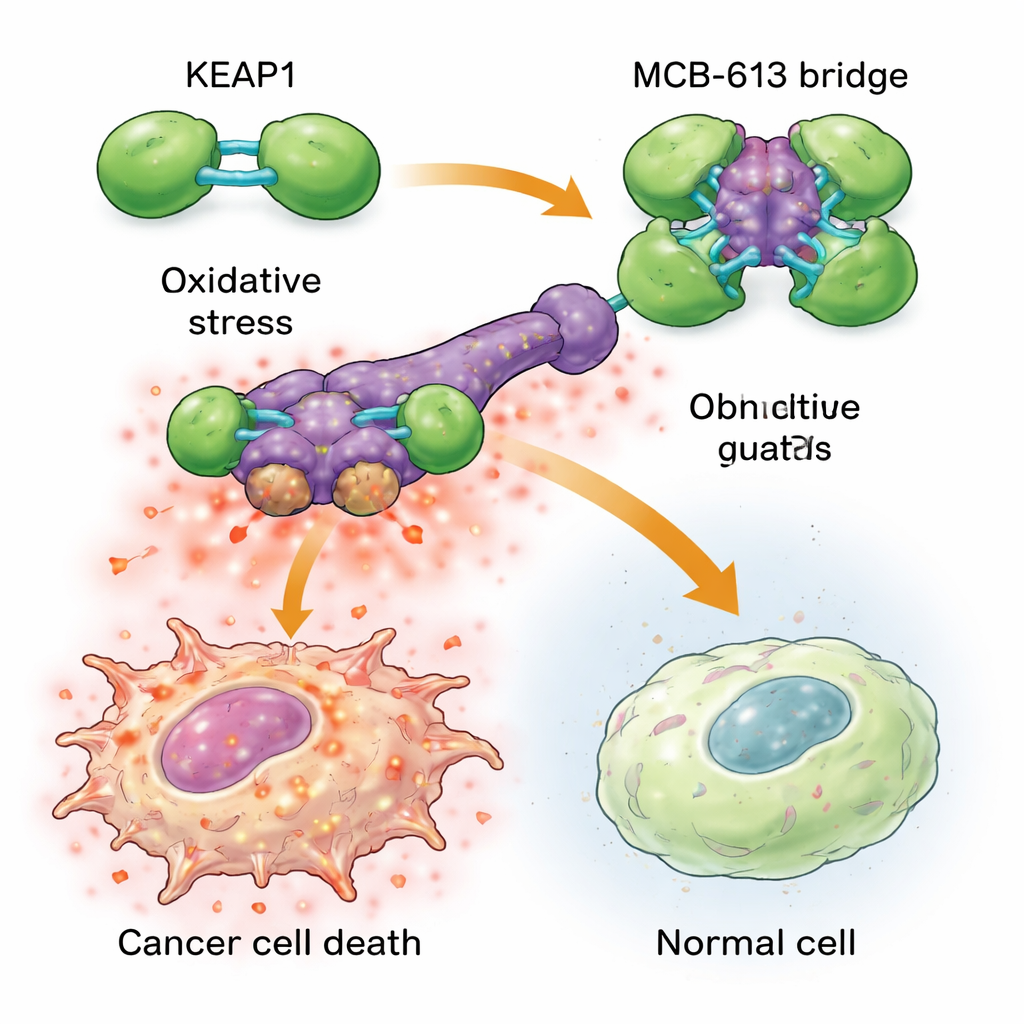
To understand why MCB-613 hits resistant cells so hard, the scientists examined which proteins it binds. Using chemical probes and a targeted CRISPR gene-cutting screen, they pinpointed a protein called KEAP1 as essential for MCB-613’s effect. KEAP1 normally acts as a cellular guardian, sensing stress and helping regulate protective responses. The team found that MCB-613 attaches to KEAP1 in an unusual way: it behaves like a rigid molecular bridge that links KEAP1 units together into oversized, abnormal clusters. This process depends not on the usual reactive sulfur-containing sites in KEAP1, but on a specific lysine amino acid in its dimerization region. When that lysine was mutated, MCB-613 could no longer clump KEAP1, and resistant cells were no longer hypersensitive to the compound.
Turning helpful stress into lethal overload Figure 2.
Clumping KEAP1 sets off a dangerous chain reaction inside drug-resistant cancer cells. These cells already live under higher background stress, with elevated levels of reactive oxygen species (damaging chemical byproducts) and increased activity in a protective signaling network known as the integrated stress response. When MCB-613 is added, KEAP1 disruption pushes this stressed state over the edge: reactive oxygen builds up further, and key stress regulators called ATF4 and CHOP switch on powerful death programs. Blocking these stress regulators, or chemically mopping up reactive oxygen, largely protected the cells from MCB-613. Interestingly, the classic KEAP1 partner NRF2, often thought of as the main driver of antioxidant defenses, was not responsible for the killing; in fact, removing NRF2 made cells even more sensitive, underscoring that MCB-613 is exploiting a different, non-canonical pathway.
What this could mean for future treatments
For now, MCB-613 itself is a tool compound with chemical drawbacks that make it unsuitable as a drug. But it reveals a powerful concept: as lung cancers evolve resistance to EGFR inhibitors, they may become locked into a stressed state that can be selectively targeted by compounds that force KEAP1 into dysfunctional assemblies. In principle, refined versions of such “molecular bridges” could be developed to be safer and more precise, giving oncologists a way to steer tumors into an “impossible choice” between sensitivity to the original targeted therapy and sensitivity to the follow-up stress-inducing agent. This evolutionary trapping strategy could eventually help delay or overcome resistance in EGFR-mutant lung cancer and perhaps other hard-to-treat cancers as well.
Citation: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge.
Nat Commun17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1