Clear Sky Science · en
Guardian ubiquitin E3 ligases target cancer-associated APOBEC3 deaminases for degradation to promote human genome integrity
Protecting our DNA from friendly fire
Our cells deploy powerful enzymes to damage viral DNA, helping us fend off infections. But some of these same enzymes, if left unchecked, can accidentally scar our own genome and contribute to cancer. This study uncovers how human cells police these risky enzymes, revealing a built-in “quality control” system that tags dangerous versions for destruction before they can rewrite our DNA.
Virus fighters that can turn against us
The APOBEC3 family of enzymes normally helps defend against viruses like HIV by chemically altering viral DNA, triggering fatal errors. Humans carry seven APOBEC3 variants, and three of them—A3A, A3B, and a form of A3H called haplotype I (A3H-I)—are strongly linked to mutation patterns seen in many cancers. These particular enzymes can move into the cell nucleus, where our chromosomes reside, and introduce characteristic clusters of mutations in the genome. Such APOBEC-linked signatures appear in over half of all human cancers, especially in breast, lung, and bladder tumors, where they expand the pool of genetic changes tumors can draw on to adapt and resist treatment.
Why the most dangerous forms are oddly unstable
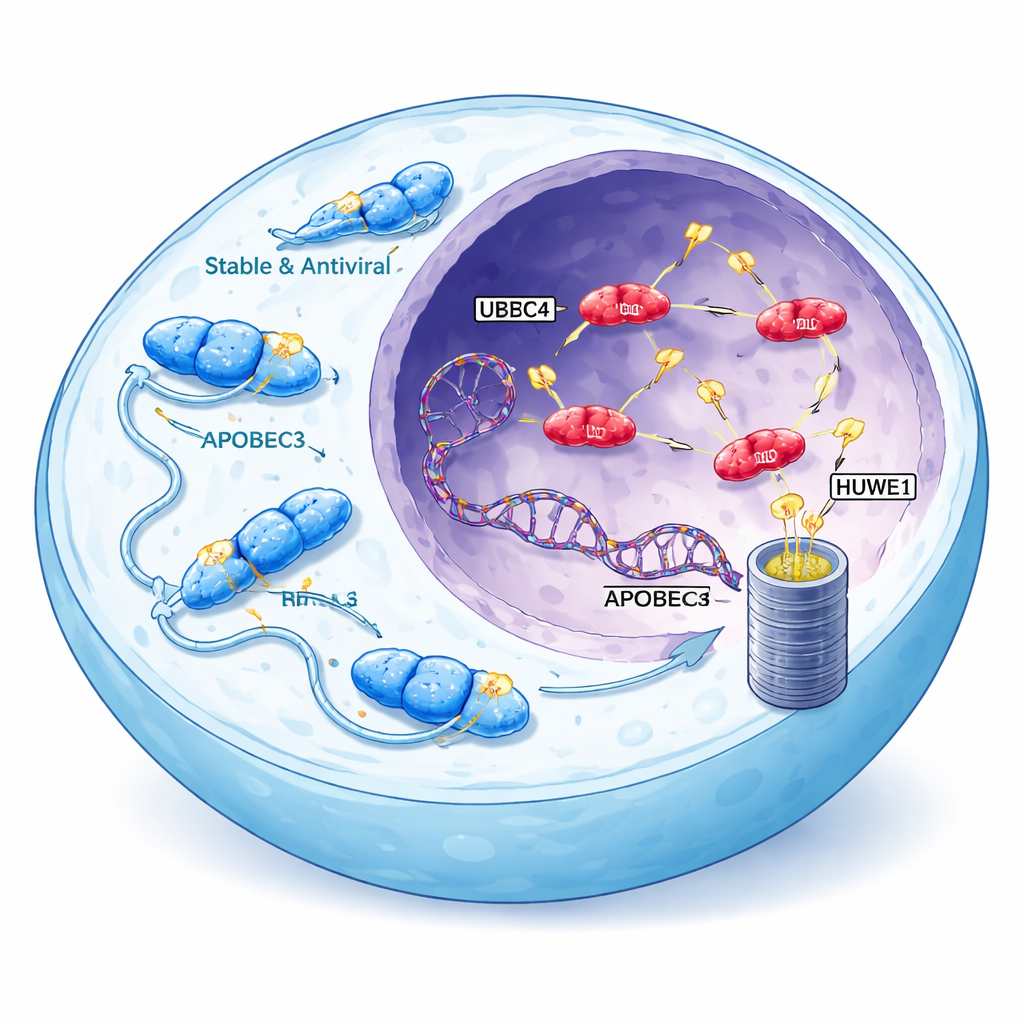
Curiously, the APOBEC3 enzymes most strongly tied to cancer are also the least stable. Unlike their cytoplasmic cousins, which accumulate to high levels and efficiently block viruses, A3A, A3B, and A3H-I are rapidly broken down and usually present at low concentrations in the nucleus. The researchers reasoned that this instability might be deliberate: if cells actively limit the levels of nuclear APOBEC3 proteins, any breakdown in that control could unleash bursts of mutagenesis. Using A3H-I as a model, they showed that cancer-associated APOBEC3s are destroyed mainly by the proteasome, the cell’s protein shredder, after being marked with small molecular “flags” called ubiquitin on multiple amino acid sites.
Finding the cellular guardians
To pinpoint the machinery responsible for tagging A3H-I and A3B, the team combined CRISPR screening with protein-proximity mapping. They engineered cells that glow differently depending on how stable A3H-I and its harmless variant A3H-II are, then systematically disrupted genes involved in protein degradation. Three ubiquitin ligases—UBR4, UBR5, and HUWE1—stood out. Knocking out any one of these ligases selectively raised levels of unstable, nuclear A3H-I without affecting the stable, cytoplasmic A3H-II. The same ligases independently boosted levels of endogenous A3B in colon and immune cell lines, and deleting all three at once produced an additive surge in A3B abundance. Biochemical experiments further showed that UBR5 and HUWE1 physically bind to A3B and A3H-I and directly attach ubiquitin chains, while UBR4 appears to specialize in extending these chains to make the proteins more efficiently disposable.
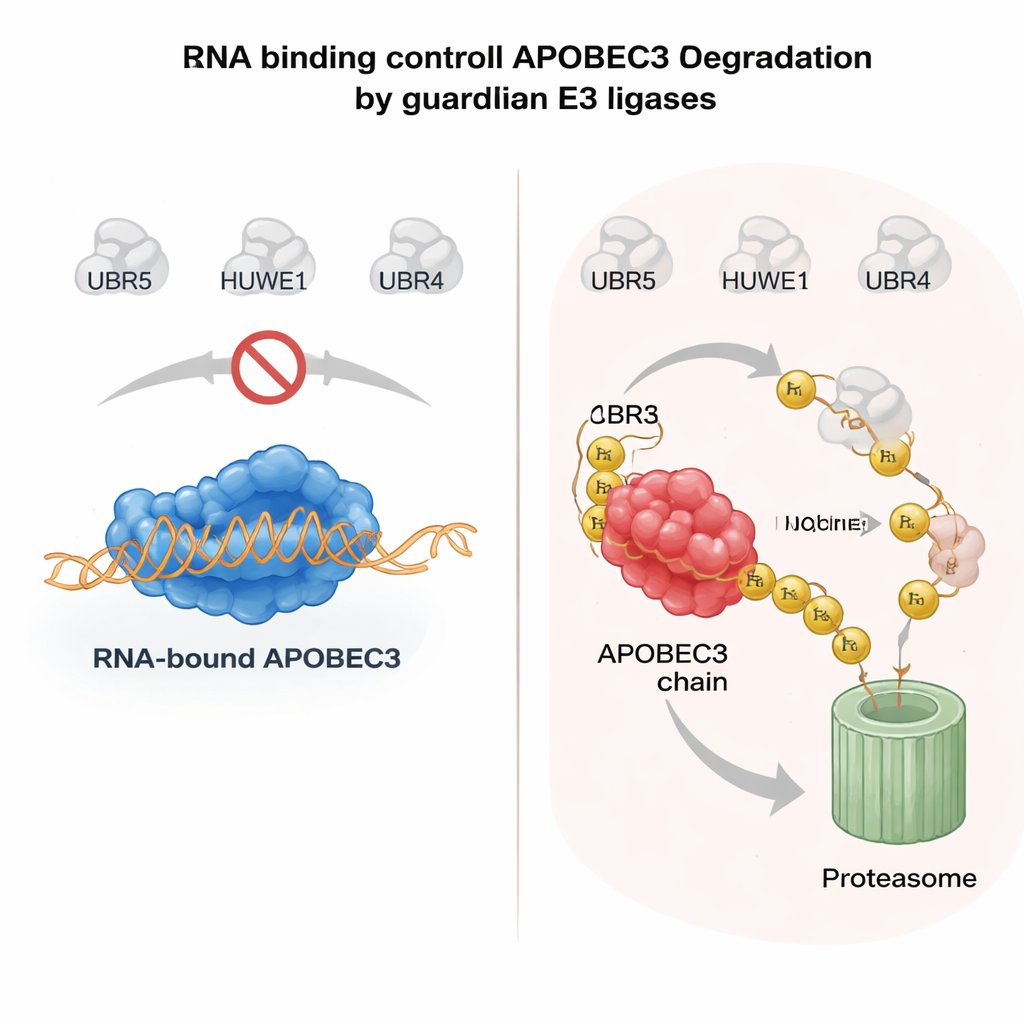
RNA as a safety belt for risky enzymes
What determines whether an APOBEC3 enzyme is spared or destroyed? A key clue came from the way APOBEC3 proteins latch onto RNA molecules in the cytoplasm. When A3H or another family member, A3G, binds RNA, it tends to stay in large complexes in the cytosol and is relatively stable. Mutations that weaken RNA binding cause these enzymes to drift into the nucleus, lose their protective partners, and become highly unstable. The authors showed that when APOBEC3 proteins are not bound to RNA, UBR5 and HUWE1 recognize exposed surfaces on the enzyme, decorate them with ubiquitin, and target them for nuclear degradation. In purified systems, adding RNase to strip away RNA greatly enhanced the ability of these ligases to bind and ubiquitinate APOBEC3, confirming that RNA binding acts as a molecular shield. This mechanism allows cells to keep antiviral APOBEC3s active in the cytoplasm while rapidly clearing any unbound, genome-threatening pool in the nucleus.
When the guardians fail: links to cancer mutations
If UBR4, UBR5, and HUWE1 act as guardians, their loss should increase mutation levels. The researchers tested this by disabling each ligase in colon cancer cells and tracking DNA changes over time using a sensitive sequencing method that reads mutational “signatures.” Removing these ligases, particularly in cells expressing A3H-I, amplified APOBEC-specific mutation patterns—exactly the kind found in human tumors. Extending this to patient data, they analyzed thousands of cancer genomes and found that tumors carrying mutations in UBR5 or HUWE1 had a significantly higher fraction of APOBEC-linked mutation signatures than tumors with intact ligases, even after accounting for overall mutation load. This suggests that defective guardian ligases in patients may allow cancer-associated APOBEC3 enzymes to run wild and reshape the tumor genome.
What this means for future cancer care
To a non-specialist, the takeaway is that our cells possess a sophisticated policing system to prevent helpful antiviral enzymes from accidentally sabotaging our own DNA. UBR4, UBR5, and HUWE1 act as guardians that sense when APOBEC3 enzymes are no longer safely bound to RNA and, especially in the nucleus, send them to the cellular shredder. When this control system is weakened—by mutations in the ligases or by drugs that block protein degradation—APOBEC3 activity can fuel the genetic chaos that drives cancer evolution and treatment resistance. Understanding this guardian network opens doors to new diagnostic markers, such as ligase mutations or APOBEC protein levels, and suggests that carefully tuning this pathway could one day help limit harmful mutation bursts in tumors without compromising our antiviral defenses.
Citation: Schwartz, I., Budroni, V., Meyenberg, M. et al. Guardian ubiquitin E3 ligases target cancer-associated APOBEC3 deaminases for degradation to promote human genome integrity. Nat Commun 17, 1723 (2026). https://doi.org/10.1038/s41467-026-68420-5
Keywords: APOBEC3, genome stability, ubiquitin ligase, cancer mutagenesis, protein degradation