Clear Sky Science · en
Comprehensive mapping of RNA modification dynamics and crosstalk via deep learning and nanopore direct RNA-sequencing
RNA’s Hidden Punctuation Marks
Our cells’ RNA molecules are not simple strings of A, C, G, and U. They are decorated with scores of tiny chemical marks that act like punctuation, helping control which genes are turned on, how proteins are made, and how cells respond to stress and disease. Yet, until now, scientists have mostly been able to study these marks one at a time, making it hard to see how they work together across the whole genome. This paper introduces ORCA, a deep‑learning system that reads native RNA molecules directly and builds a global, multi-layered map of these chemical marks and how they interact.
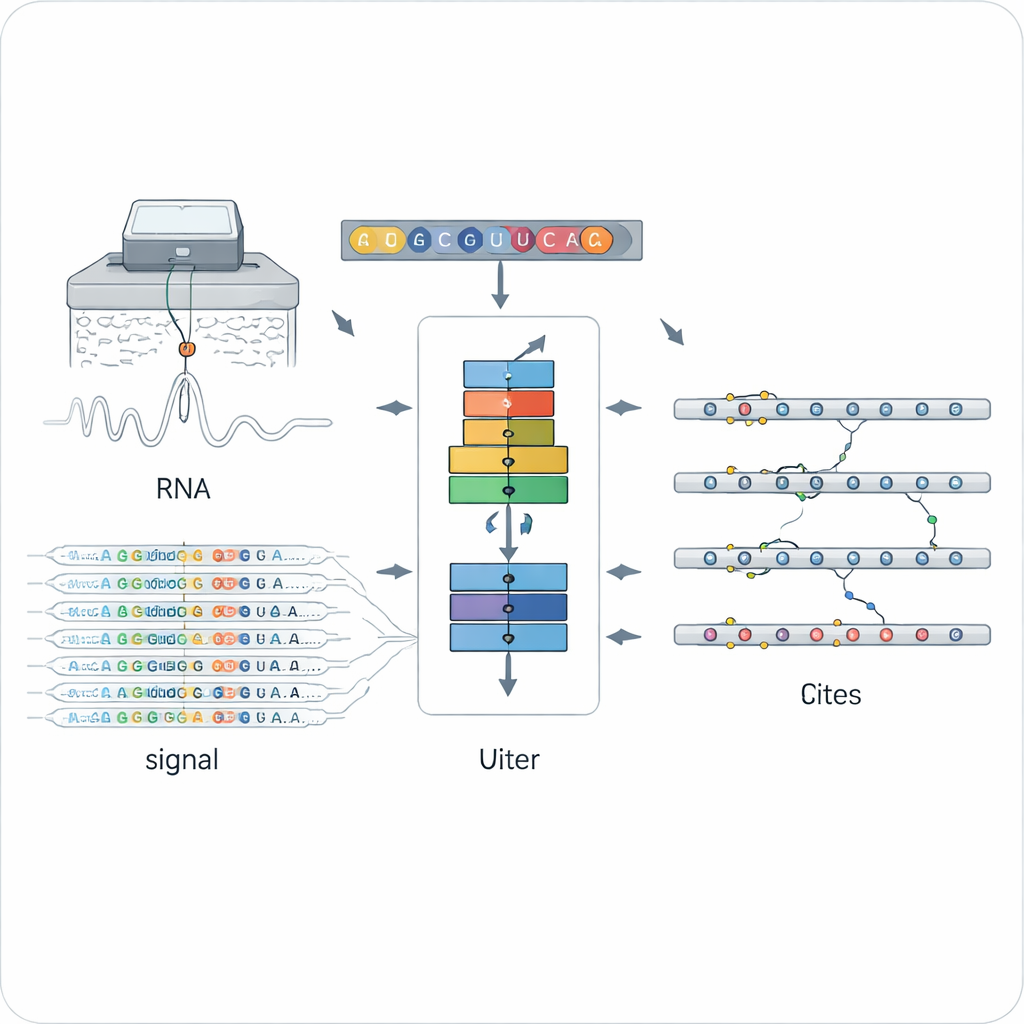
A New Way to Read Chemical Marks on RNA
Traditional methods for finding RNA modifications usually rely on special antibodies or chemistry that are tuned to a single type of mark, such as the popular N6‑methyladenosine (m6A). That makes them powerful but narrow: each method sees only one kind of mark, often in one experimental setup. Nanopore direct RNA sequencing opened a different door by threading individual RNA molecules through a tiny pore and measuring changes in electrical current that depend on the exact chemical structure of each base. Modified and unmodified letters distort the signal and basecalling in subtly different ways, but making sense of this noisy, high‑dimensional data across many modification types has been a major challenge.
Teaching a Neural Network to Spot Any Mark
ORCA (Omni‑RNA modification Characterization and Annotation) tackles this challenge in two stages. First, it focuses on a small window around every position in the RNA and aggregates both the raw electrical signal and the pattern of sequencing errors across many reads. Because only a fraction of RNA copies carry a given mark, truly modified sites show more skewed signal distributions and more frequent basecalling mistakes at that position. ORCA uses a deep recurrent neural network trained with an “adversarial” strategy so that it learns general patterns that distinguish modified from unmodified sites, without locking onto any single known chemical type. This allows ORCA to assign each site a modification score and an estimated fraction of molecules that are modified.
Learning the Identity of Each Mark
In the second stage, ORCA learns to label what kind of chemical mark is present. The authors feed the model a set of high‑confidence sites from public databases, where conventional experiments have already identified m6A, 5‑methylcytosine (m5C), pseudouridine (Ψ), inosine, 2′‑O‑methylation, and several rarer marks. ORCA compresses the signal patterns, sequence context, and short sequence “motifs” around each site into a lower‑dimensional map, then fine‑tunes itself to predict the modification type and the exact base it sits on. Crucially, unlabelled sites are also used as “background” examples, which helps the model avoid forcing unknown marks into the wrong category. Once trained, ORCA can transfer these learned labels to tens of thousands of previously unannotated sites across the transcriptome.
Seeing Many Modifications at Once
Applying ORCA to human and mouse cells, the authors show that it not only matches or exceeds the accuracy of leading tools for specific marks like m6A, m5C, and Ψ, but can also detect marks it was never explicitly trained on. For example, even when m6A data were withheld during training, ORCA recovered most independently measured m6A sites and correctly distinguished them from similar sequence motifs that are unmodified. It did the same for 2′‑O‑methyl groups, inosine editing sites, and a wide variety of chemical changes on ribosomal RNA, including many rare modifications measured by mass spectrometry. Overall, ORCA greatly expands the known catalog of RNA modification sites, with multi‑fold increases in annotated m5C, Ψ, m7G, and other low‑abundance marks compared with existing databases.
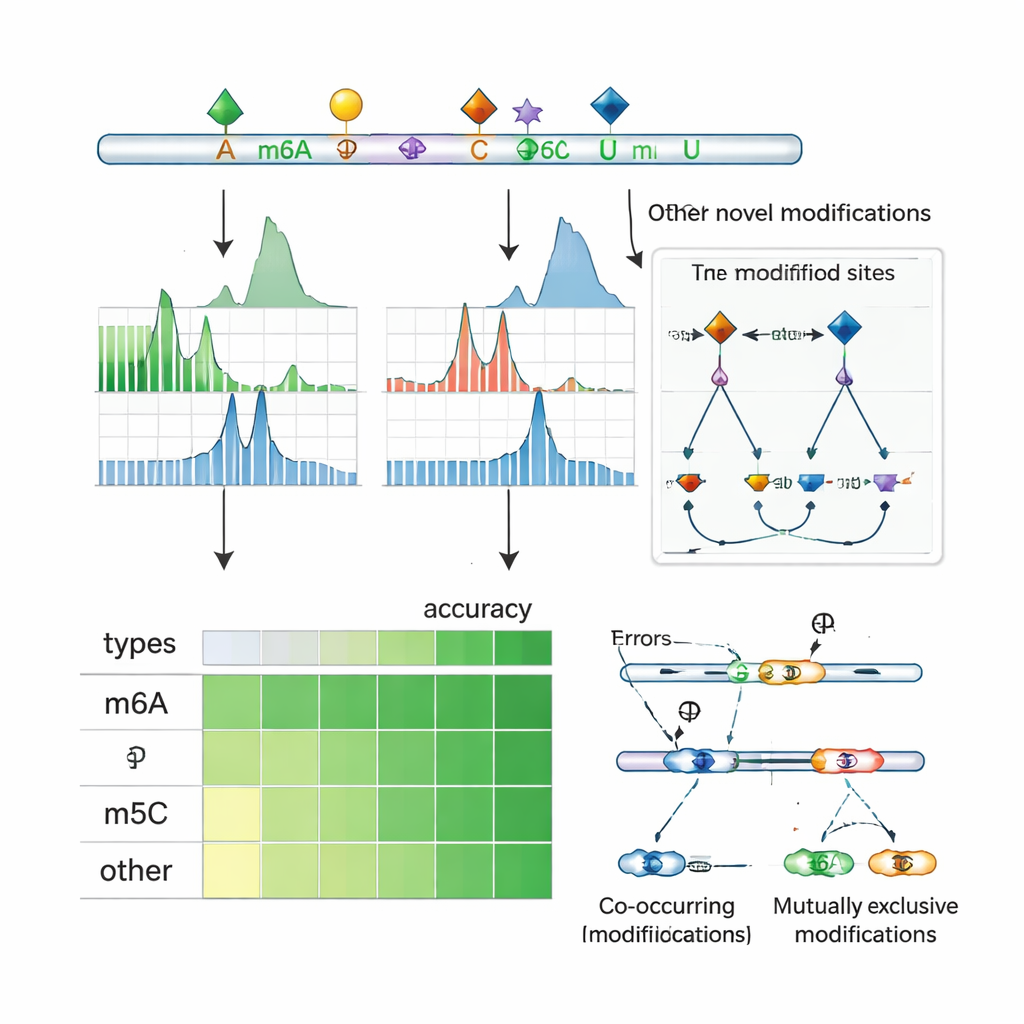
Uncovering Crosstalk and Splicing Control
Because nanopore sequencing reads entire RNA molecules, ORCA can examine which marks appear together on the same transcript and which tend to exclude each other. The authors cluster nearby marks along RNAs and use a probabilistic model to infer whether pairs of sites are often co‑modified or mutually exclusive on single molecules. They find frequent co‑occurrence of m6A with m5C and other marks, as well as many regions where one site is modified only when the neighboring site is not. In human cell lines, these patterns often fall near exons that are alternatively included or skipped, and they overlap binding sites for splicing regulators and “reader” proteins that recognize modified RNA. In specific genes, ORCA reveals that certain splice variants are enriched for one pattern of marks, while alternative variants carry a different pattern, linking local chemical decoration of RNA to how messages are cut and stitched together.
Why This Matters for Biology and Medicine
By combining direct RNA sequencing with flexible deep learning, ORCA turns a complicated electrical signal into a rich, multi‑layered map of chemical marks across the transcriptome. For non‑specialists, the key outcome is that scientists can now see not just where individual RNA modifications occur, but how many different marks decorate the same molecule and how those combinations relate to gene regulation, especially RNA splicing. This framework makes it possible to study RNA “epigenetics” in many cell types and conditions without designing a new experiment for each mark, paving the way for discoveries about how these tiny chemical tweaks contribute to development, brain function, and diseases such as cancer and neurological disorders.
Citation: Dong, H., Gao, Y., Cai, Z. et al. Comprehensive mapping of RNA modification dynamics and crosstalk via deep learning and nanopore direct RNA-sequencing. Nat Commun 17, 1722 (2026). https://doi.org/10.1038/s41467-026-68419-y
Keywords: RNA modifications, nanopore sequencing, deep learning, epitranscriptome, alternative splicing