Clear Sky Science · en
Using the linear references from the pangenome to discover missing autism variants
Why hidden DNA changes matter for autism
Most families who pursue genetic testing for an autistic child hope for clear answers, but roughly four out of five receive no definitive genetic explanation. This study tackles a key reason why: many impactful DNA changes are too complex for standard tests to see. By building nearly complete genomes for 189 people from 51 autism-affected families and comparing them to a new, richer "pangenome" reference, the researchers show how advanced sequencing can uncover rare, previously invisible mutations that may help explain some cases of autism and related conditions.
Looking beyond standard genetic tests
Traditional clinical testing relies on short DNA snippets to scan a person’s genome. This works well for many single-letter changes but often fails in repetitive or structurally complex regions, exactly where some powerful disease-causing mutations hide. The team focused on families in which earlier short-read genome, exome, or gene panel tests had not found a cause for autism or Rett-like symptoms. Using long-read sequencing, which reads much larger stretches of DNA, they built high-quality, phased genome assemblies for 189 individuals. This means they could reconstruct each person’s two copies of every chromosome, one inherited from each parent, with very few gaps.
Structural variants: big changes with big effects
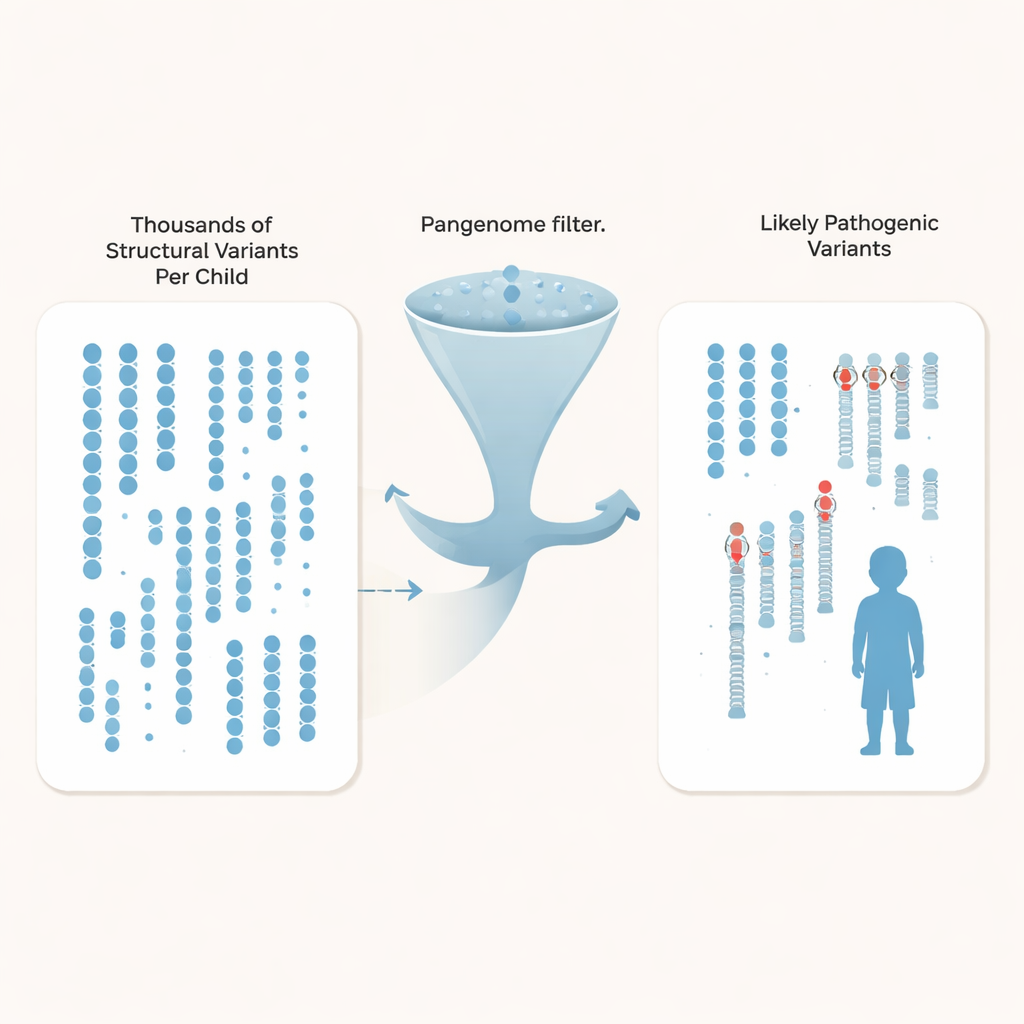
Rather than just tracking single-letter differences, the researchers zeroed in on structural variants—insertions, deletions, and rearrangements that affect at least 50 DNA letters and can disrupt genes or their control switches. Each child carried around 27,000 such variants, but the vast majority are harmless background differences shared across the population. By comparing their autism families against hundreds of deeply sequenced pangenome control genomes drawn from diverse ancestries, the team could filter out over 97% of common structural variants for each child, leaving roughly 600 rare candidates per genome, and as few as about 200 when using the largest control set.
Finding missed mutations in known risk genes
With the search space drastically reduced, the authors integrated several lines of evidence: known autism and neurodevelopmental disorder genes, regulatory regions active in the developing human cortex, and patterns of inheritance within each family. They uncovered three clearly pathogenic mutations that earlier tests had missed. These included a new stop signal in the SYNGAP1 gene, which is important for synapse function, and a deletion that chops off the last exon of MECP2, a key Rett syndrome gene, even though the patient had undergone multiple prior clinical tests. They also confirmed a disease-causing change in TBL1XR1, a gene that interacts with MECP2. In total, they highlighted nine additional structural variants—often inherited and located in regulatory regions near brain-related genes—as strong candidates for future functional testing.
What the study did not find—and why that still matters
Despite this deep search, the authors did not see a clear overall excess of structural variants in autistic children compared with their unaffected siblings, at least in this modest sample size. There was, however, a hint of more structural changes on the X chromosome in affected girls, and the near-complete X and Y assemblies allowed them to spot unusual patterns such as extreme skewing of X-chromosome inactivation. These features could become important clues as more families are studied. Crucially, the work shows that long-read sequencing can recover pathogenic variants that short-read methods miss, especially in tricky parts of the genome and in the control regions that fine-tune gene activity.
What this means for families and future testing
For families, the immediate impact is modest but meaningful: among these difficult-to-solve cases, about 6% received a clear genetic diagnosis, and nearly one in five gained strong new candidate variants to investigate. For the field, the message is larger. As more diverse, complete reference genomes are added to the pangenome and long-read sequencing becomes more accessible, clinicians will be able to rule out common structural changes and focus quickly on a small set of rare, potentially harmful variants in each patient. That shift could gradually turn today’s many “unsolved” autism cases into ones where the underlying biology—and possible paths to support and treatment—are far better understood.
Citation: Sui, Y., Lin, J., Noyes, M.D. et al. Using the linear references from the pangenome to discover missing autism variants. Nat Commun 17, 1681 (2026). https://doi.org/10.1038/s41467-026-68378-4
Keywords: autism genetics, long-read sequencing, structural variants, human pangenome, Rett syndrome