Clear Sky Science · en
DOT1L activity limits transcription elongation velocity and favors RNAPII pausing to facilitate mutagenesis by AID
How our immune cells fine-tune risky DNA edits
Our immune system creates potent antibodies by deliberately mutating its own DNA, a risky strategy that can sometimes fuel cancer. This study asks a deceptively simple question with big implications: what controls where, and how efficiently, these intentional mutations occur? The answer centers on a protein called DOT1L, which adjusts the speed of gene reading inside B cells and, in doing so, helps aim the mutation machinery at the right spots.
Turning on mutation to sharpen antibodies
When B cells encounter an infection, they upgrade their antibodies in two ways. They introduce tiny changes in the antibody’s binding region to improve how tightly it grabs germs, and they swap out the antibody’s tail to change how the immune system responds. Both upgrades start with an enzyme called AID, which nicks and alters DNA in actively read genes. While AID is crucial for good immunity, it can also hit other genes, creating dangerous breaks that drive blood cancers. Earlier work showed that AID prefers genes that are heavily read and regulated by powerful DNA switches called super-enhancers, but this did not fully explain why only a select subset of genes are truly vulnerable.
A chromatin marker that flags AID-sensitive genes
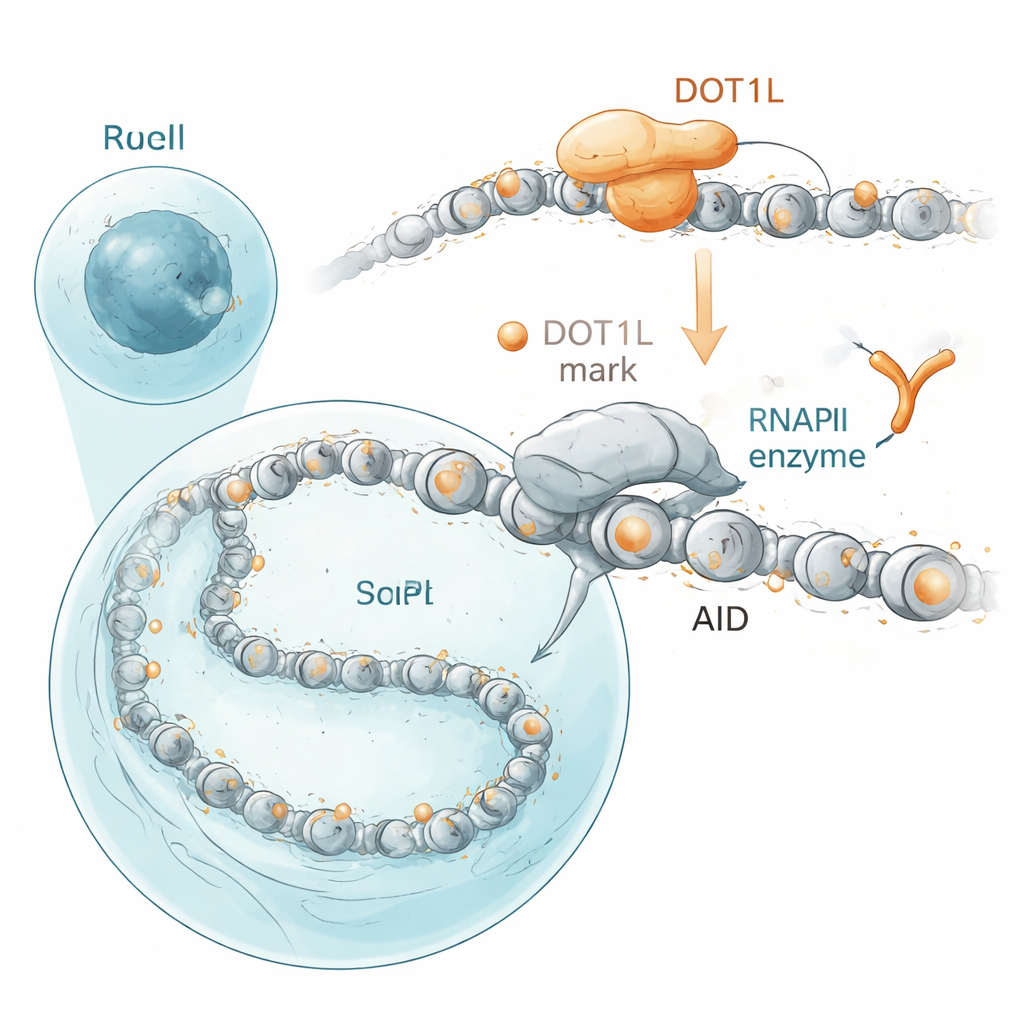
The authors homed in on proteins that sit close to AID in the nucleus. Using a proximity-labeling technique in human cells, they found that AID clusters near DOT1L, an enzyme that decorates a specific spot on the histone proteins around which DNA is wrapped. This decoration, a chemical tag on histone H3 at position K79, is common on active genes. In mouse B cells, genes that AID frequently mutates—including antibody genes and cancer-linked off-targets—carry particularly high levels of these DOT1L-made marks. When the researchers disabled DOT1L in B cell lines or blocked its activity with a drug, antibody “class switching” dropped, and so did AID-driven DNA breaks and cancer-prone fusions between antibody genes and the growth gene cMyc. Importantly, DOT1L’s catalytic function, not just its presence, was required: mutant versions that could no longer place the histone mark failed to restore normal antibody switching.
Slowing the gene reader to give AID time
At first glance, this is puzzling because DOT1L is associated with active genes, yet removing it did not simply shut genes off. Using a method that snapshots newly made RNA, the team discovered that B cells lacking DOT1L actually produced more nascent transcripts across many DOT1L-marked genes—even though there was slightly less of the main gene-reading enzyme, RNA polymerase II, sitting on those genes. By combining this nascent RNA readout with maps of polymerase occupancy, they inferred that, under normal conditions, DOT1L’s histone marks act like gentle speed bumps. They slow the polymerase as it moves along the gene and extend short pauses near the start and within the body of DOT1L-marked genes. Without DOT1L, the polymerase runs faster and pauses for shorter times. Because AID needs brief windows when DNA is exposed and the polymerase lingers, this speed-up paradoxically reduces AID’s ability to latch on and do its job, even as overall transcription goes up.
Decoupling gene activity from mutation risk
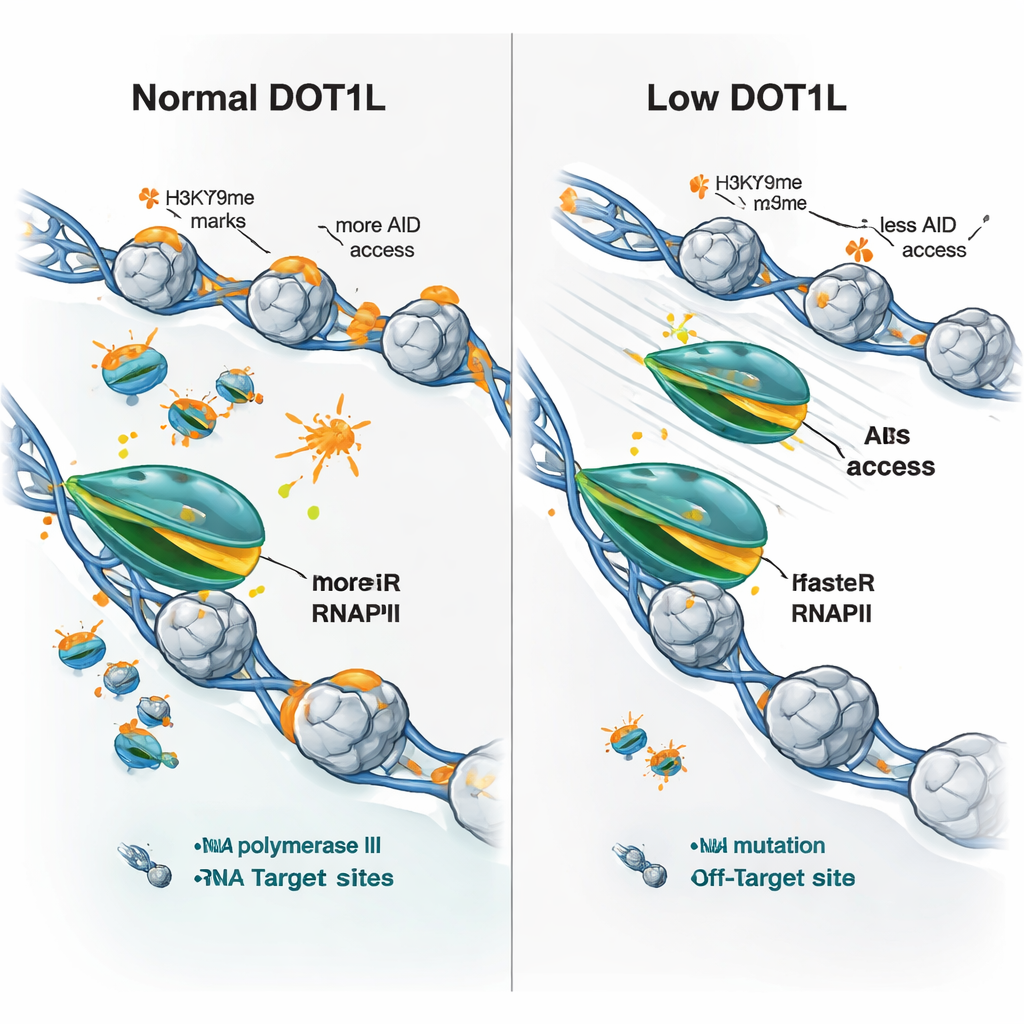
The researchers then asked whether these speed changes could account for the mixed pattern of gene expression seen when DOT1L is lost—some genes go up, others go down. They found that virtually all DOT1L-marked genes shared one feature in knockout cells: faster elongation by RNA polymerase II. But the outcome depended on the starting state. Slow, weakly expressed genes tended to produce more RNA when the polymerase sped up, while highly active, long genes with already fast polymerases sometimes produced less, likely because too-rapid passage disrupts efficient processing or completion. Crucially, at both antibody genes and classic AID off-targets, DOT1L loss led to faster polymerase movement, less evidence of polymerase “stalling,” and significantly reduced AID occupancy, even when the genes themselves were not turned down.
Why this matters for immunity and cancer
Taken together, the work paints DOT1L as a subtle traffic controller for the machinery that reads genes in B cells. By installing specific histone marks, DOT1L slightly slows RNA polymerase II and extends its pauses, creating a transcriptional environment where AID can productively engage antibody genes—and, unfortunately, a limited set of other vulnerable genes—to introduce mutations. When DOT1L is missing or inhibited, the polymerase speeds through, leaving AID with fewer chances to act, which blunts antibody diversification and simultaneously lowers the risk of certain harmful rearrangements. This mechanistic insight explains why DOT1L loss can both raise and lower gene expression, and it links the fine control of transcription speed directly to where our immune system dares to rewrite its own DNA.
Citation: Subramani, P.G., Seija, N., Ridani, J. et al. DOT1L activity limits transcription elongation velocity and favors RNAPII pausing to facilitate mutagenesis by AID. Nat Commun 17, 1623 (2026). https://doi.org/10.1038/s41467-026-68332-4
Keywords: antibody diversification, AID enzyme, DOT1L, gene transcription, B cells