Clear Sky Science · en
Molecular basis of antagonism of the dimeric human arginine vasopressin receptor 1A
Why a hormone brain receptor matters
Hormones such as vasopressin and oxytocin are best known for controlling water balance, blood pressure, childbirth, and bonding. But the way their receptors work at the atomic level has remained largely hidden. This paper reveals detailed 3D structures of one key receptor, the human vasopressin V1a receptor, which is linked to social behavior, stress, and several brain disorders. Understanding its shape and how drugs block it could help scientists design better treatments for conditions like autism, post-traumatic stress disorder, and Huntington’s disease.
A twin receptor that shapes heart, kidney, and brain signals
The V1a receptor sits on the surface of many cells in the body, especially in blood vessels, kidneys, and certain brain regions. When the hormone vasopressin docks onto it, the receptor switches on internal signaling pathways that control blood pressure, fluid balance, and brain circuits for social interaction, emotion, and stress. Genetic and clinical studies have tied abnormal V1a signaling to autism spectrum disorder, PTSD, and Huntington’s disease, making it an attractive drug target. Several V1a-blocking drugs (antagonists) are already in use or in clinical trials, but until now no one had seen the human V1a receptor at high resolution, leaving major questions about how it assembles and how exactly these drugs shut it down. 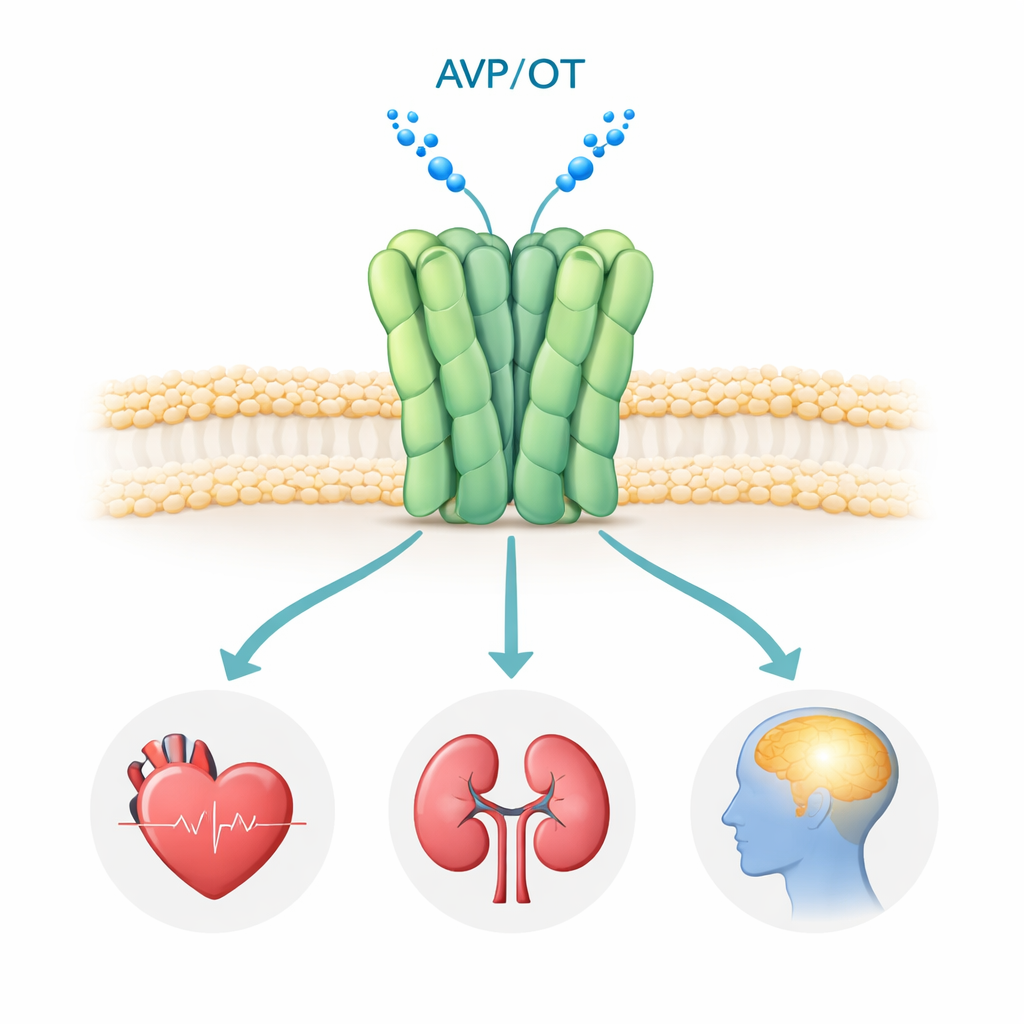
Capturing the receptor’s structure in multiple drug-bound states
The researchers used cryo–electron microscopy, a technique that flash-freezes proteins and images them with an electron beam, to visualize the human V1a receptor. To stabilize the protein, they engineered a slightly modified form that still binds drugs well and paired it with a small antibody fragment (a nanobody) to aid imaging. They solved structures of the receptor alone and bound to three medically important antagonists: atosiban (a peptide drug used to prevent premature labor), and two brain-penetrant small molecules, balovaptan and SRX246, which have been tested in people with autism or Huntington’s disease. All structures reached near-atomic resolution, revealing the positions of the receptor’s seven membrane-spanning helices, flexible loops, and the bound drugs.
A receptor that prefers to work in pairs
Unlike related receptors that had been seen only as single units, V1a appeared as a pair—a dimer—in all four cryo-EM structures. The two receptors lie side by side in the membrane, making tight contacts mainly through one of their helices, helped by both polar and greasy (hydrophobic) contacts. To test whether this pairing also occurs in living cells, the team fused V1a to a bright fluorescent protein and used a single-molecule photobleaching method: if a spot on the cell surface contained two receptor copies, its light would disappear in two steps. About three-quarters of the observed spots bleached in exactly two steps, strongly supporting the idea that V1a naturally forms dimers at the cell surface. When the scientists mutated key contact residues to disrupt the interface, the receptor shifted toward single units and became less responsive to hormone and drug, implying that the dimer is not just structural decoration but functionally important. 
A flexible gate at the hormone entryway
The team uncovered an unexpected “gate” region called extracellular loop 2 (ECL2), which sits at the top of the hormone-binding pocket. In the drug-free (apo) state, this loop lies flat across the pocket like a lid and does not form the usual disulfide bond (a sulfur–sulfur link) seen in many related receptors. Instead, parts of the loop fold into the pocket and are held in place by a web of interactions with surrounding helices, partially covering the large, sticky binding cavity. When any of the three antagonists binds, ECL2 swings up and away, forms the classic disulfide bond, and creates a wide, solvent-filled cavity that the drugs occupy. This dramatic motion suggests that V1a may use ECL2 as a dynamic barrier to limit random activation by stray molecules, and that drugs could be designed either to trap the loop in its flat “ground state” or to exploit its raised, open conformation.
How three drugs silence the same receptor in different ways
Atosiban, which closely mimics the natural hormone oxytocin, stretches from the top of the pocket to its base, anchoring itself via a combination of hydrogen bonds and hydrophobic contacts. By changing a few key positions compared with oxytocin, it fails to trigger the chain of internal shifts normally required for receptor activation: crucial “microswitch” residues that move during signaling stay locked in their inactive positions, the internal cavity that admits the G protein never opens, and a magnesium-binding site important for activation is disrupted. In contrast, balovaptan and SRX246 are compact, non-peptide molecules that burrow deeply into the pocket but use distinct strategies. Balovaptan relies on a rigid, hydrophobic core that packs tightly into a deep cleft, plus a flexible polar tail that reaches toward the pocket entrance. SRX246 uses a modular, fragment-like architecture anchored by a β-lactam core, with different “zones” plugging subpockets and extending toward the extracellular loops. In both cases, the drugs stabilize an inactive conformation that is incompatible with G protein binding. Subtle differences in pocket shape and chemistry—especially at two positions on helices 5 and 7—help explain why balovaptan and SRX246 prefer V1a over closely related receptors.
Implications for future therapies
By providing high-resolution snapshots of V1a as a dimer, revealing a previously unseen “flat” loop conformation in the drug-free state, and detailing how three very different antagonists shut the receptor down, this work gives drug designers a precise structural map for targeting V1a. It suggests ways to craft next-generation medicines that either exploit dimer-specific features or lock the receptor into an especially inactive ground state, with the ultimate aim of treating brain and stress-related disorders more specifically and with fewer side effects.
Citation: Zhong, P., Chu, B., Yu, Z. et al. Molecular basis of antagonism of the dimeric human arginine vasopressin receptor 1A. Nat Commun 17, 1622 (2026). https://doi.org/10.1038/s41467-026-68331-5
Keywords: vasopressin V1a receptor, G protein–coupled receptor, receptor dimerization, cryo-EM structure, neuropsychiatric drug design