Clear Sky Science · en
Computational design of generalist cyclopropanases with stereodivergent selectivity
Why tiny three‑ring structures matter for medicines
Cyclopropanes—three‑membered carbon rings—are tiny, strained building blocks that can dramatically change how a drug behaves in the body. The exact 3D arrangement of atoms (its stereochemistry) can decide whether a molecule becomes a useful medicine or an inactive or even harmful look‑alike. This paper describes a computational strategy to design enzymes that can reliably make all four possible 3D forms of these rings from the same starting materials, opening the door to faster, cleaner exploration of drug candidates.
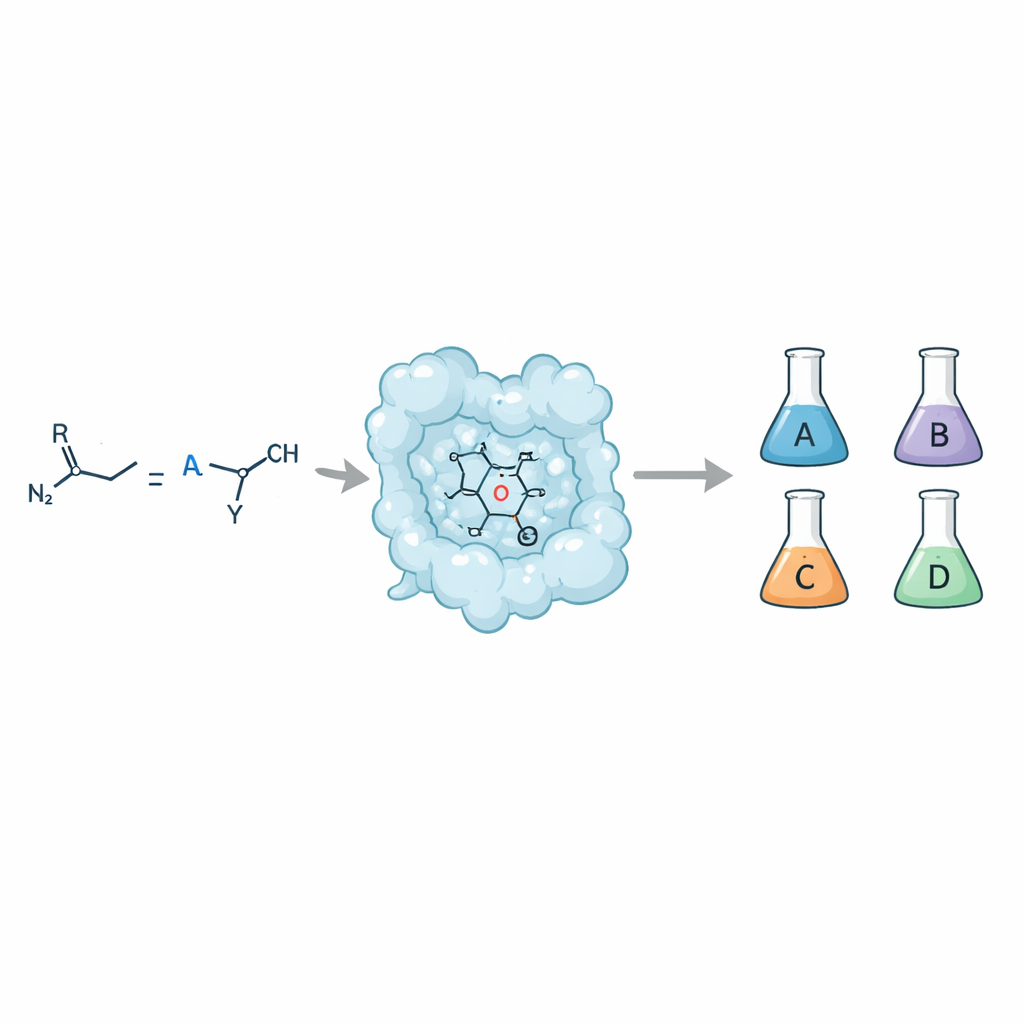
From one recipe to four different outcomes
When a simple double bond (an olefin) reacts with a carbene donor such as a diazo compound, the result can be a cyclopropane ring. But that ring can adopt four distinct stereoisomeric forms, each with the same atoms but arranged differently in space. Chemists want access to every one of these forms because they can interact very differently with biological targets and influence key drug properties such as absorption, metabolism, and safety. Traditional small‑molecule catalysts can sometimes deliver this control, but doing so with enzymes—nature's own catalysts—has been difficult, especially when aiming for both high selectivity and tolerance of many different substrates.
Designing enzymes on a computer screen
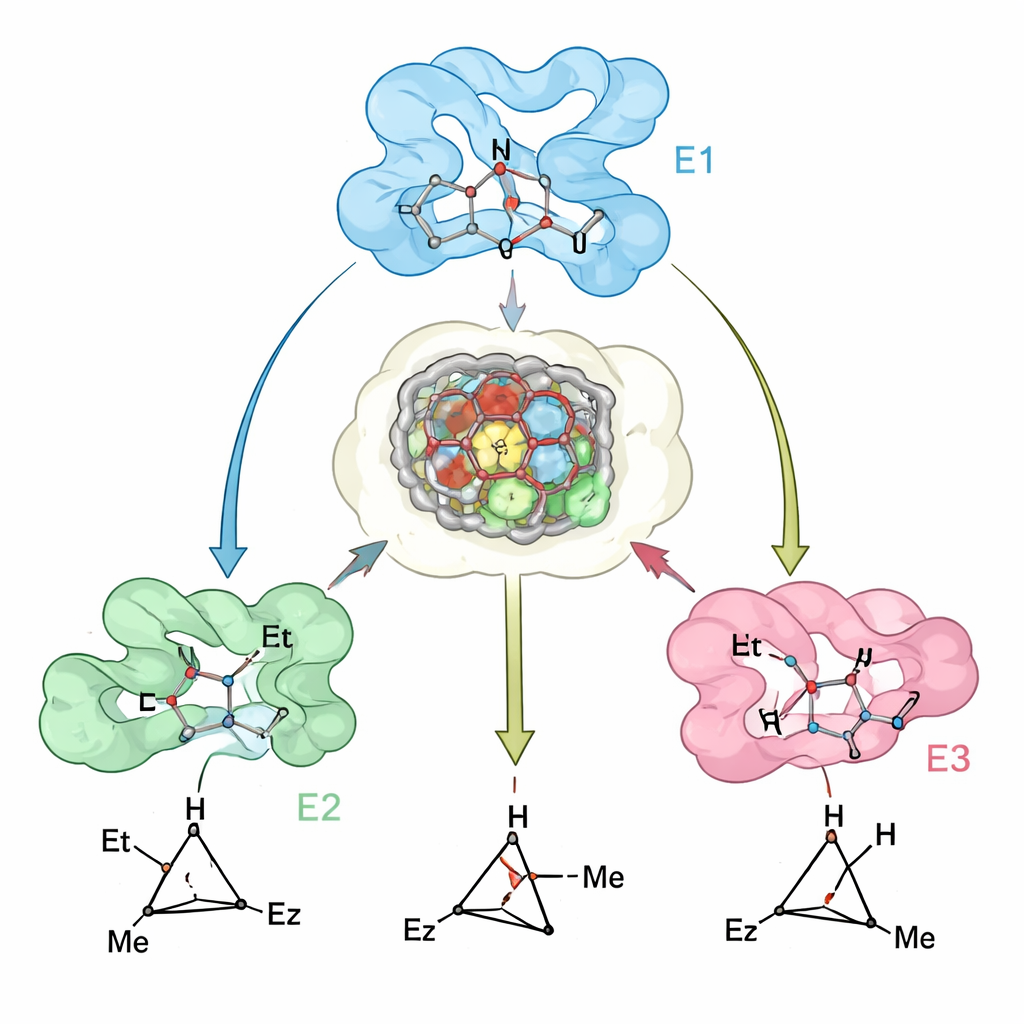
The authors developed a mechanism‑based, multi‑state computational workflow to solve this problem. They first used quantum chemical calculations to model the fleeting transition states—the high‑energy structures along the reaction path—for forming each of the four cyclopropane stereoisomers. These models were then placed into the active sites of different heme‑containing proteins, and the Rosetta protein design software was used to evaluate how well each protein stabilized or destabilized each transition state. Crucially, the design score rewarded mutations that both favored the desired transition state (positive design) and disfavored the competing ones (negative design), effectively teaching the enzyme to "prefer" one 3D product over the others.
Building a complete enzyme toolbox
Using this approach, the team created a family of "generalist" cyclopropanase enzymes. Starting from myoglobin, they redesigned its active site to obtain variants that produce the trans-(1R,2R) cyclopropane with very high selectivity and good activity across more than 20 different olefins, including demanding unactivated and electron‑poor substrates. A previously engineered myoglobin already supplied the complementary trans-(1S,2S) product. To reach the two cis products, the authors turned to other heme proteins. They remodeled the bacterial enzyme P450cam to obtain variants that selectively give the cis-(1S,2R) product, and they repurposed human indoleamine 2,3‑dioxygenase‑1 (IDO1)—not previously used for carbene chemistry—to favor the cis-(1R,2S) product. Altogether, these four biocatalysts can deliver every stereoisomer of the same cyclopropane product set, often with up to 99% control of both diastereomer and enantiomer.
Seeing how design matches reality
To test how well their computational models reflected real enzymes, the researchers solved crystal structures of a key myoglobin variant and compared them with the predicted structures. The agreement was close, and the experimental data highlighted a subtle but important feature: the protein active site is pre‑organized to welcome the preferred transition state, while small shifts in nearby loops and helices make binding of the "wrong" transition state energetically unfavorable. Where the predictions were less accurate—such as for a few bulky substrates—the discrepancies could be traced to backbone movements that were not fully captured in the modeling, suggesting clear paths for improving future design methods.
What this means for future drugs and catalysts
By combining physics‑based transition state modeling with smart protein redesign, this work demonstrates that stereochemical outcomes of enzyme‑catalyzed reactions can be programmed in advance, rather than discovered only through trial‑and‑error evolution. The resulting suite of cyclopropanases offers chemists a practical way to make complete sets of cyclopropane stereoisomers from a wide range of starting olefins, greatly simplifying structure–activity studies in drug discovery and natural product synthesis. The same strategy should be adaptable to other enzyme types and reaction classes, accelerating the creation of biocatalysts that give precise 3D control over complex molecules.
Citation: Shen, Z., Siriboe, M.G., Ren, X. et al. Computational design of generalist cyclopropanases with stereodivergent selectivity. Nat Commun 17, 1620 (2026). https://doi.org/10.1038/s41467-026-68327-1
Keywords: biocatalysis, cyclopropanation, enzyme design, stereochemistry, heme proteins