Clear Sky Science · en
De novo variants in the splicing factor gene SF3B1 are associated with neurodevelopmental disorders
When One Gene Disrupts the Brain’s Early Blueprint
Why do some children develop learning difficulties, seizures, or feeding problems even when pregnancy and birth seem normal? This study examines a single gene, called SF3B1, that helps cells process genetic messages. The researchers show that new, spontaneous changes in this gene can subtly scramble how brain cells read DNA instructions, leading to a previously unrecognized neurodevelopmental syndrome.
A Master Editor of Genetic Messages
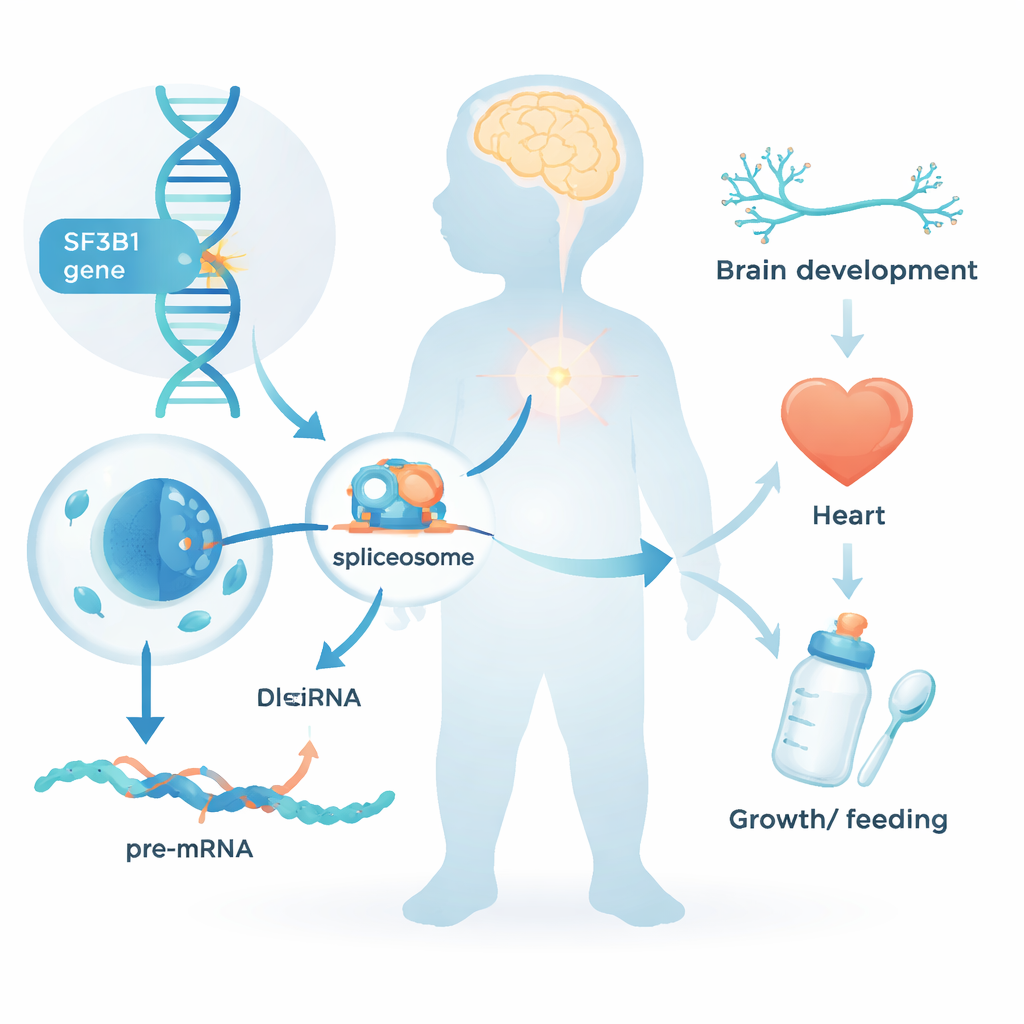
Every cell in our body must convert raw genetic text into clear instructions before it can make proteins. This editing step, known as RNA splicing, removes non‑coding segments and stitches together the useful pieces. SF3B1 is a central component of the cellular “splicing machine.” Until now, changes in SF3B1 were mainly known for their role in cancers, where tumor cells acquire mutations in this gene during life. The new work asks a different question: what happens when a damaging change in SF3B1 is present from conception in every cell of the body?
A Newly Recognized Childhood Syndrome
The team gathered data from 26 children and young adults who all carried rare SF3B1 variants, mostly arising de novo—that is, not inherited from either parent. Nearly all had neurodevelopmental problems: delays in learning to sit, walk, or talk; intellectual disability of mostly mild to moderate degree; and, in about half of the cases, seizures. Many had low muscle tone and required extra help with feeding, sometimes through a gastric tube. Facial features were subtly unusual but not identical from child to child; a strikingly common trait was a high or cleft palate. Several participants also had heart defects, growth restriction, or small head size, showing that the impact of SF3B1 changes extends beyond the brain.
Two Classes of Genetic Changes, Two Clinical Patterns
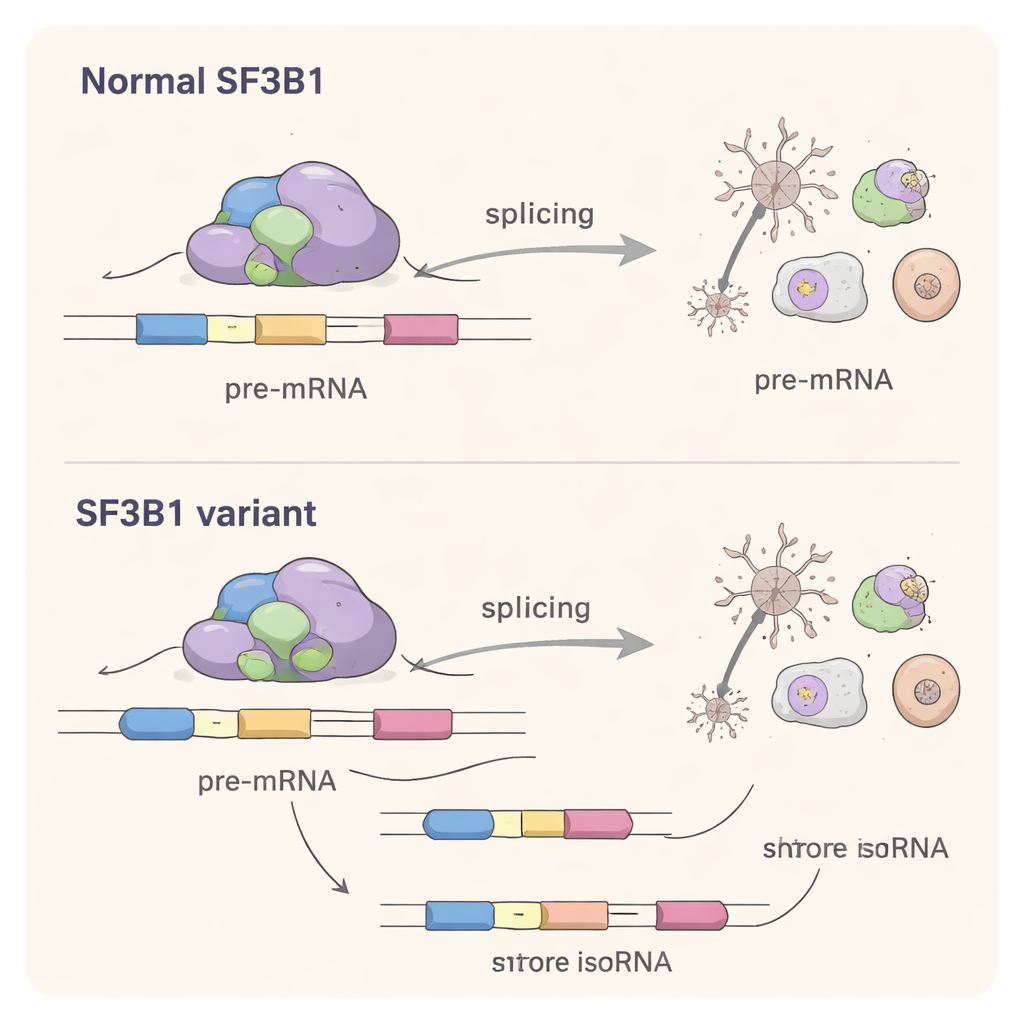
The researchers distinguished between two broad types of SF3B1 variants. One group included “loss‑of‑function” changes, such as premature stop signals, that are expected to reduce the amount of working SF3B1 protein. The second group contained missense variants, where a single amino acid in the protein is altered. By clustering the children’s medical features, the team observed that those with missense variants tended to have more severe and more complex problems, including a higher rate of heart and gastrointestinal anomalies, short stature, and microcephaly. Loss‑of‑function variants, in contrast, were sometimes inherited from a mildly affected or even apparently healthy parent, suggesting that simply having less SF3B1 can be compatible with relatively mild symptoms in some individuals.
Fine-Tuning Errors Rather Than a Complete Breakdown
To understand what the missense variants do inside cells, the scientists recreated them in laboratory cell lines. Surprisingly, these altered SF3B1 proteins could still perform the basic job of splicing well enough to rescue cells in which the normal SF3B1 had been silenced. This ruled out a simple loss‑of‑function explanation. Using deep RNA sequencing, the team then looked across the entire set of cellular messages. They found that missense variants subtly shifted the splicing of hundreds of genes, especially by changing which splice sites were chosen at the ends of exons and by causing occasional exon skipping. The scale of disruption was smaller than that seen with the classic cancer‑associated SF3B1 mutation K700E, but still substantial: many affected genes are involved in brain development, nerve wiring, and fundamental processes like RNA handling and protein synthesis.
A Shared Mechanism Between Cancer and Brain Disorders
Although most of the neurodevelopmental SF3B1 variants occur at different positions from the well‑known cancer mutations, they disturb the same core process: precise recognition of splice sites in RNA. The study shows that these developmental variants have their own “splicing signature,” choosing alternative splice sites that are often closer to the normal ones than those favored in cancer. This suggests a change‑of‑function mechanism, where the mutated protein competes with the normal copy and nudges the splicing machinery toward slightly wrong choices in many genes at once.
What This Means for Families and Future Research
For affected families, the work identifies SF3B1 as a new cause of neurodevelopmental disorders that can now be tested in genetic clinics, potentially ending long diagnostic searches. More broadly, it adds SF3B1 to a small but growing list of splicing genes whose alterations can drive both cancer and childhood brain disorders, depending on when and how the gene is changed. By mapping how specific SF3B1 variants reshape RNA splicing, the study lays the groundwork for future therapies aimed at correcting mis-splicing in a targeted way.
Citation: Uguen, K., Bergot, T., Scott-Boyer, MP. et al. De novo variants in the splicing factor gene SF3B1 are associated with neurodevelopmental disorders. Nat Commun 17, 1569 (2026). https://doi.org/10.1038/s41467-026-68284-9
Keywords: RNA splicing, SF3B1, neurodevelopmental disorders, de novo variants, spliceosomopathies