Clear Sky Science · en
Molecular basis for the inhibition of de novo DNA methylation by TCL1A
How Cells Decide What to Remember
Every cell in your body carries essentially the same DNA, yet brain cells, blood cells, and skin cells behave very differently. One way cells "remember" their identity is through chemical tags added to DNA, a process called DNA methylation. This study uncovers, at atomic detail, how a small protein called TCL1A can switch off the enzymes that write those methyl marks. Because both DNA methylation and TCL1A are linked to cancer and reproductive disorders, understanding this molecular tug‑of‑war could eventually inspire new therapies.
The Cell’s DNA Tagging Machinery
DNA methylation works like a pencil mark in the genome’s margins, helping silence some genes and stabilize the genome as cells develop. Two enzymes, DNMT3A and DNMT3B, are the main "writers" that place new methyl tags during early development and when stem cells specialize. If these enzymes are mutated or misregulated, the pattern of DNA marks can be scrambled, contributing to developmental syndromes and blood cancers. TCL1A is a protein best known for its role in immune cell cancers, where it is frequently overproduced. Earlier work hinted that TCL1A can bind DNMT3A and DNMT3B and blunt their activity, but no one knew exactly how it achieves this block.
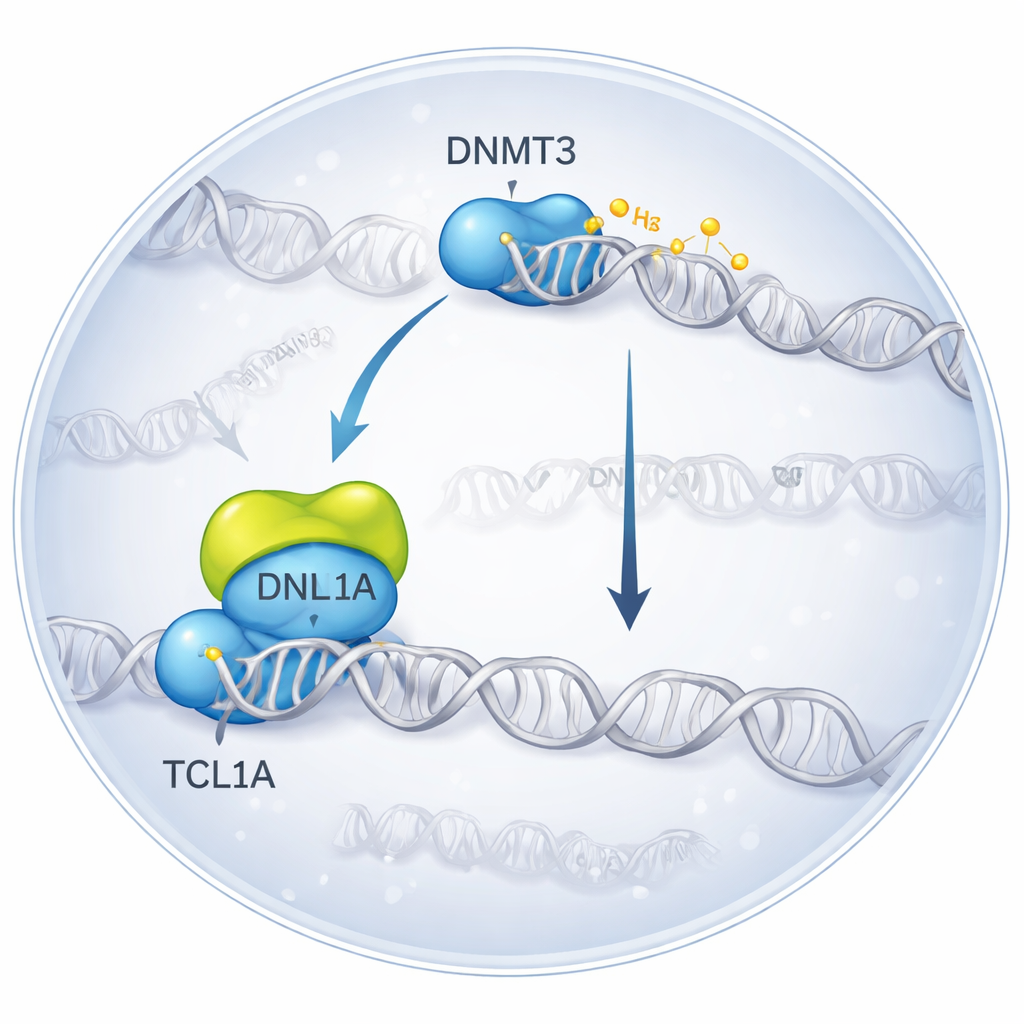
Freezing a Molecular Encounter in 3D
The researchers used cryo‑electron microscopy, a technique that images flash‑frozen molecules, to visualize the complex formed when DNMT3A binds TCL1A. They found that two DNMT3A molecules pair up, and on each side a TCL1A dimer docks onto the catalytic portion of DNMT3A—the very region that normally interacts with helper proteins and DNA. This binding surface overlaps with the spot where another partner, DNMT3L, usually attaches to boost DNMT3A’s activity. In biochemical tests, adding TCL1A sharply reduced the ability of both DNMT3A and DNMT3B to methylate DNA, even in the presence of DNMT3L, confirming that the structural complex corresponds to a strongly inhibited state.
A Shape Shift That Jams the Enzyme
Looking more closely, the team saw that TCL1A binding does not simply sit on top of the active site like a lid. Instead, it triggers a subtle but far‑reaching change in DNMT3A’s shape. Two flexible regions of the enzyme, known as the target‑recognition loop and the catalytic loop, swing away from the positions they occupy when DNMT3A is bound to DNA. In the active form, these loops nestle against DNA and form a pocket for a small molecule fuel called SAM, which donates the methyl group. With TCL1A attached, the catalytic loop instead folds into the SAM pocket and blocks it, while also making it harder for DNA to reach the enzyme. Binding measurements confirmed that DNMT3A partnered with TCL1A can no longer grip DNA or SAM detectably.
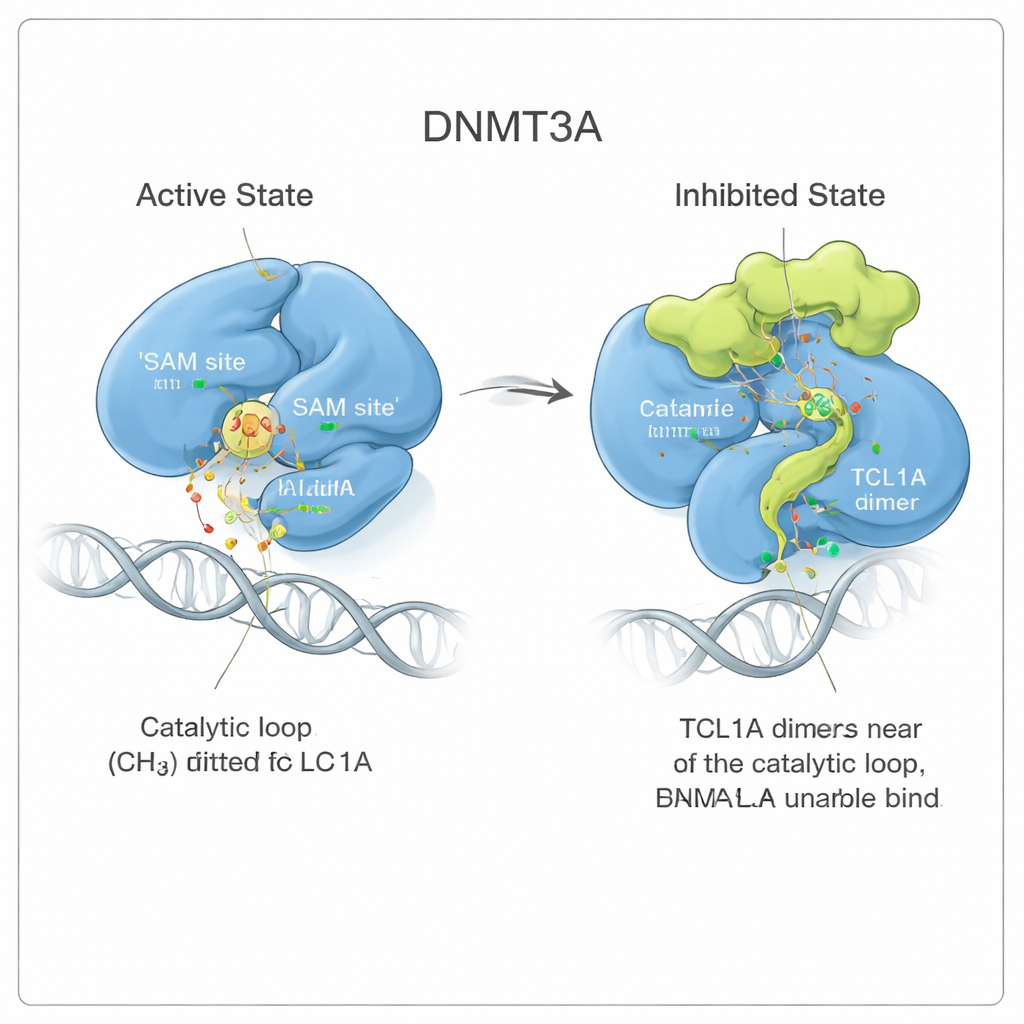
Watching a Dynamic Block in Motion
To understand how stable this inhibited conformation is, the authors ran long molecular dynamics simulations, essentially physics‑based movies of the molecules in solution. When DNMT3A was bound to its activator DNMT3L, the catalytic loop stayed snug in its active position. When TCL1A was present, that loop became much more mobile, flapping around but repeatedly occupying the SAM pocket like seaweed swirling yet still clogging a drain. This constant motion shrank the available space for SAM more than ten‑fold, supporting a model in which TCL1A exploits DNMT3A’s natural flexibility to impose a dynamic, rather than rigid, form of inhibition.
Consequences for Developing Cells and Disease
The team then asked what this molecular blockade means for real cells. They engineered mouse embryonic stem cells to produce human TCL1A during a stage when the cells normally ramp up DNA methylation as they begin to differentiate. Genome‑wide methylation mapping showed that cells overexpressing TCL1A failed to gain the usual high level of DNA methylation, closely resembling cells in which both Dnmt3a and Dnmt3b genes were knocked out. A mutant version of TCL1A that binds DNMT3 enzymes poorly had little effect, underscoring that the physical interaction is key. These findings link the structural mechanism to broad epigenetic changes across the genome.
What This Means for Health
Taken together, the work reveals how TCL1A can act as a powerful brake on the enzymes that lay down new DNA methylation marks. By docking at a critical interface, TCL1A repositions flexible loops in DNMT3A and DNMT3B so that they can no longer bind their DNA template or chemical fuel, leading to a global loss of methyl tags in cells. In normal development, this kind of fine‑tuned control may help balance when and where methylation is added. When TCL1A is misplaced or overproduced, as in certain blood cancers and rare reproductive disorders, that same mechanism can derail the cell’s epigenetic program. Understanding this interaction at atomic resolution opens the door to designing molecules that mimic or counteract TCL1A’s effects, potentially restoring healthy DNA methylation patterns.
Citation: Liu, Q., Li, J., Wang, X. et al. Molecular basis for the inhibition of de novo DNA methylation by TCL1A. Nat Commun 17, 2159 (2026). https://doi.org/10.1038/s41467-025-67710-8
Keywords: DNA methylation, DNMT3A, TCL1A, epigenetics, cancer