Clear Sky Science · en
Reduction of RAD23A extends lifespan and mitigates pathology in a mouse model of TDP-43 proteinopathy
Why this research matters to families and patients
Many forms of dementia and motor neuron disease, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), involve proteins in brain cells that misfold, clump, and slowly poison neurons. One of the key culprits is a protein called TDP-43, which normally helps manage RNA but becomes toxic when it aggregates. This study asks a hopeful question: can we make brain cells more resilient by dialing down another protein, RAD23A, that helps control how damaged proteins are handled? The authors show in mice that lowering RAD23A can extend life, improve movement, and lessen brain damage in a model of TDP-43–driven disease, hinting at a new kind of treatment strategy.
A protein traffic jam in sick neurons
Neurodegenerative diseases are often marked by piles of misfolded proteins that the cell’s waste-disposal machinery fails to clear. In ALS and FTD, TDP-43 moves out of the nucleus, forms sticky clumps, and becomes heavily tagged with ubiquitin, a signal that normally directs proteins to the proteasome, the cell’s main shredder. RAD23A is one of several “shuttle” proteins that can carry ubiquitin-tagged cargo to the proteasome. Yet earlier work in worms and cultured neurons suggested that losing RAD23-like proteins could actually protect against TDP-43–induced damage, a paradox that this study set out to explore in a living mammalian brain.
Turning down RAD23A in a TDP-43 mouse model
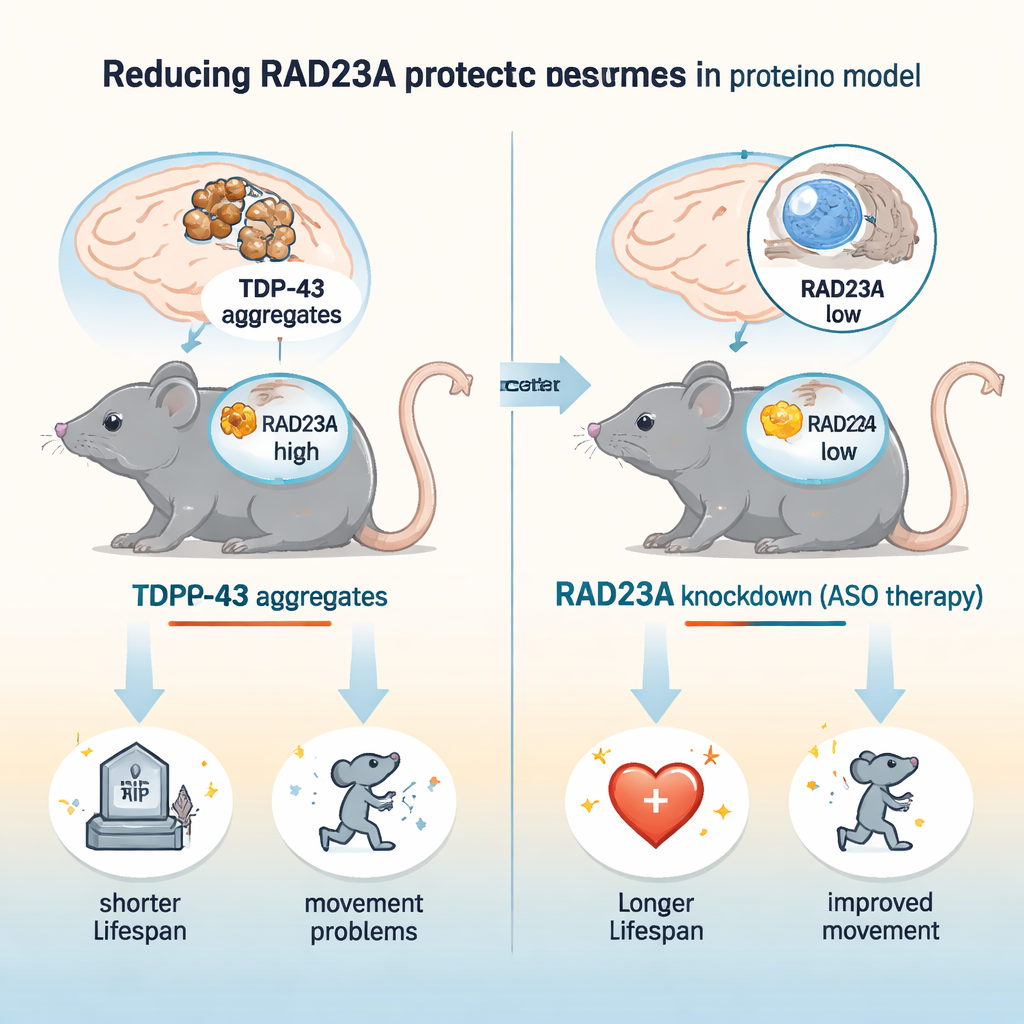
The researchers used a well-established mouse model, called TAR4/4, that overproduces human TDP-43 in neurons and develops movement problems, spinal curvature, tremor, and early death, mirroring key features of ALS/FTD. They lowered RAD23A in two ways: by injecting newborn mice with antisense oligonucleotides (ASOs) that reduce Rad23a RNA, and by breeding mice that carry a genetic knockout of Rad23a. A single ASO treatment cut RAD23A levels in brain and spinal cord by about three quarters. In these TDP-43 mice, RAD23A knockdown extended lifespan by roughly 50% and delayed the onset and severity of gait problems, tremor, spinal curvature, and hindlimb clasping. Interestingly, a complete genetic loss of RAD23A did not add further benefit, suggesting that partial reduction is optimal and that long-term, total absence may trigger compensatory changes.
Less inflammation, cleaner protein handling, and a calmer genome
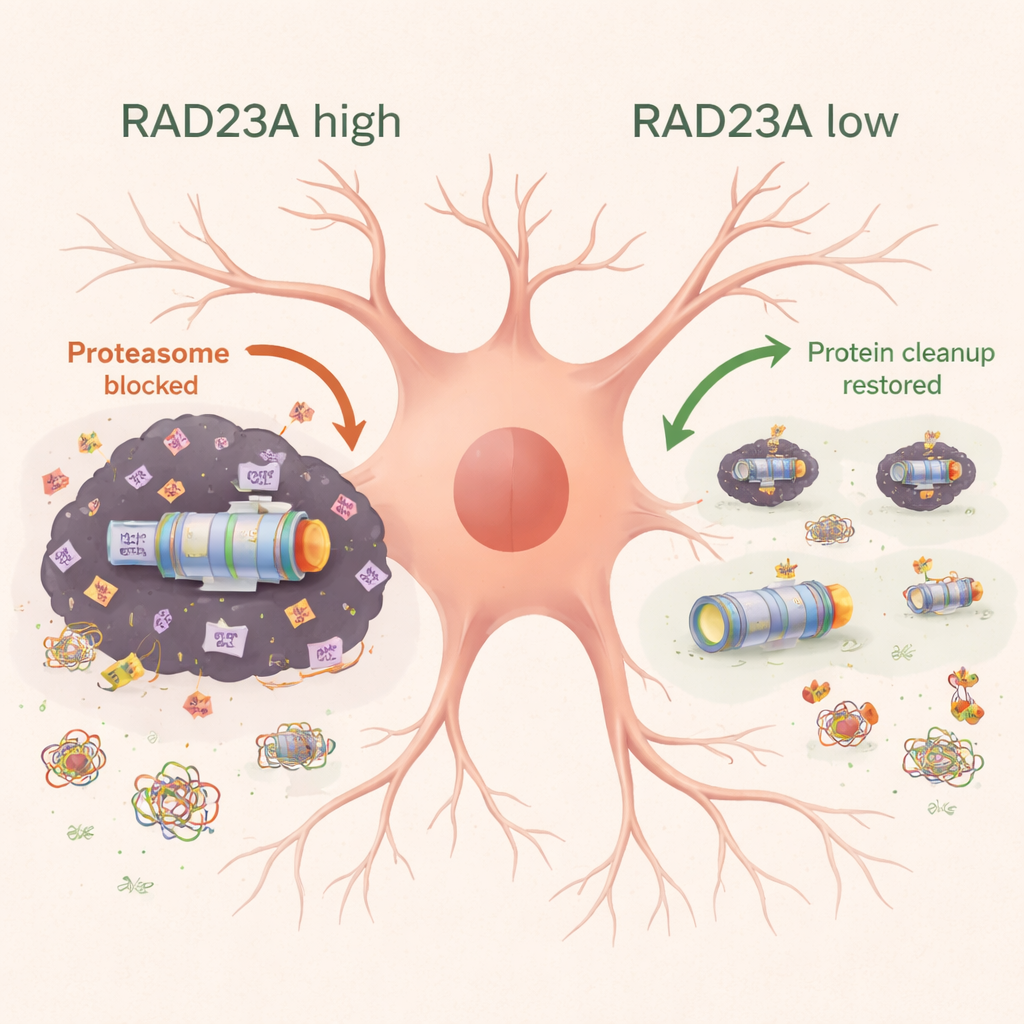
Microscopic examination of the motor cortex showed that TDP-43 mice lost neurons and developed strong activation of astrocytes and microglia, the brain’s support and immune cells. Lowering RAD23A preserved neuron numbers and reduced markers of inflammation and cell death. Biochemical analyses revealed that TDP-43 overproduction flooded cells with ubiquitin-tagged, detergent-insoluble proteins and dragged proteasome subunits into these aggregates, weakening the cell’s ability to clear damaged proteins. Reducing RAD23A lowered the total load of ubiquitinated proteins, kept more proteasomes in the soluble, working pool, and restored several types of proteasome activity toward normal. At the same time, RAD23A knockdown reduced both total and aggregated forms of TDP-43, including a particularly toxic 25-kilodalton fragment, and shifted TDP-43 away from the cytoplasm back toward the nucleus. Genome-wide RNA sequencing showed that thousands of gene expression changes triggered by TDP-43 were partially reversed when RAD23A was reduced, especially genes involved in neuronal function, mitochondrial energy production, and aggregate-clearing pathways such as aggrephagy.
Remodeling the hidden “insoluble” proteome
To look more closely at the stubborn aggregates that resist normal detergents, the team used heavy-isotope mass spectrometry to catalog proteins trapped in the insoluble fraction of mouse cortex. Expression of human TDP-43 drew in proteasome components, cytoskeletal and transport proteins, and other cellular machinery. When RAD23A was knocked down, the overall composition of this insoluble proteome shifted: fewer proteasome and transport-related proteins were sequestered, while some ribosomal and stress-related proteins increased in the aggregates. Notably, this remodeling did not simply mirror changes in RNA levels, suggesting that RAD23A mainly influences how existing proteins are partitioned between soluble and aggregated states, rather than how much of each protein is made.
What this could mean for future therapies
Together, these findings paint RAD23A as a powerful tuner of protein quality control in neurons under stress. By partially lowering RAD23A in a TDP-43–driven mouse model, the authors were able to reduce toxic protein clumps, restore the activity of the protein-disposal machinery, quiet harmful gene-expression changes, limit brain inflammation, and prolong life and motor function. Because abnormal TDP-43 accumulation is widespread in both inherited and sporadic forms of ALS, FTD, and related disorders, targeting RAD23A with human-compatible antisense drugs might offer a way to protect neurons without directly blocking TDP-43 itself, an essential protein. While much remains to be tested in other models and in humans, this work identifies RAD23A as a promising new handle on a common pathway of neurodegeneration.
Citation: Guo, X., Prajapati, R.S., Chun, J. et al. Reduction of RAD23A extends lifespan and mitigates pathology in a mouse model of TDP-43 proteinopathy. Nat Commun 17, 1820 (2026). https://doi.org/10.1038/s41467-025-65104-4
Keywords: TDP-43, ALS, protein aggregation, proteasome, antisense therapy