Clear Sky Science · en
Loss-of-function variants in ODAD1 disrupt ODA docking and induce actin cytoskeletal remodeling in primary ciliary dyskinesia
When the Body’s Microscopic Brushes Go Wrong
Every breath you take depends on armies of tiny, hair‑like structures called cilia that sweep mucus, germs, and dust out of your airways. In people with a rare inherited condition called primary ciliary dyskinesia, or PCD, these microscopic brushes don’t work properly, leading to stubborn coughs, lung infections, and sometimes reversed organ placement in the body. This study uncovers how damaging changes in a single gene, ODAD1, not only disable the cilia’s internal motors but also unexpectedly rewire the scaffolding of the cells that host them—revealing a new, potentially druggable weak point in the disease.
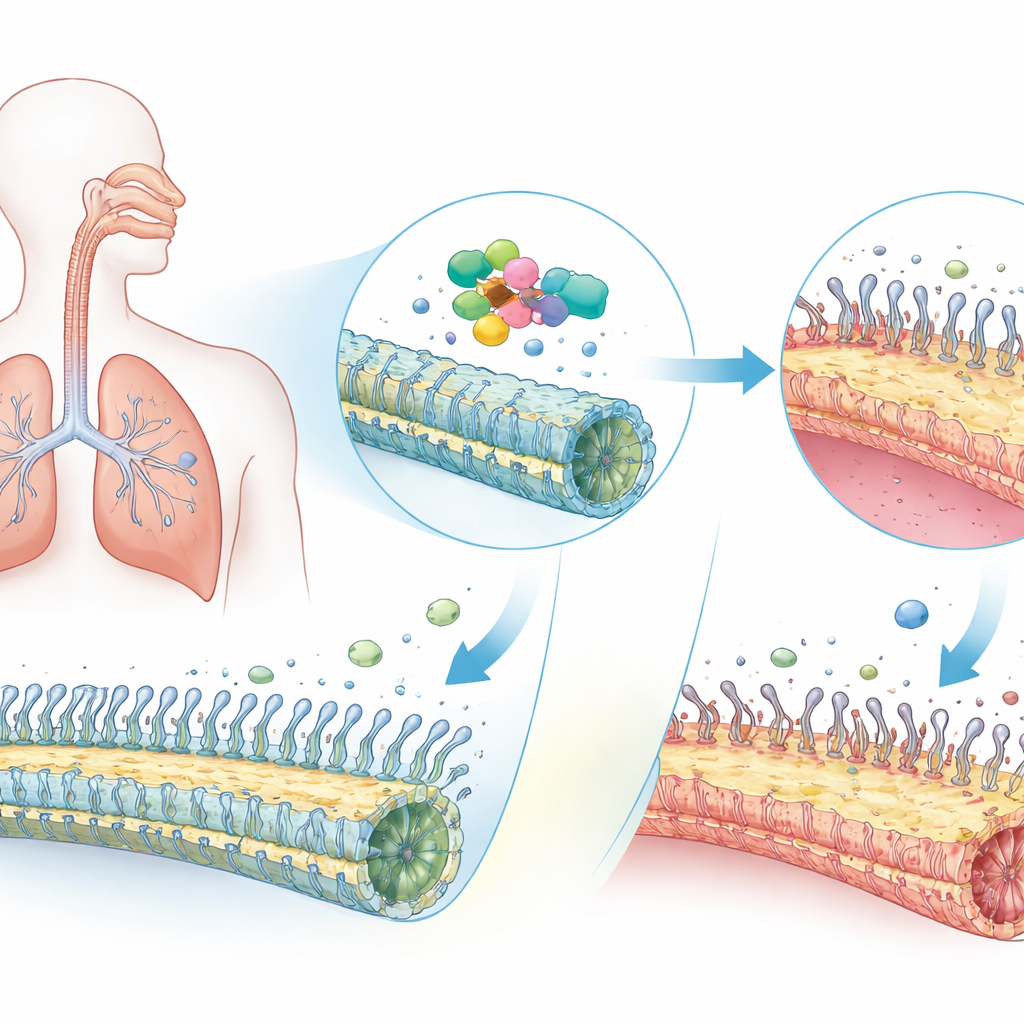
A Rare Lung Disease with Big Consequences
PCD affects about one in several thousand people worldwide and often appears in childhood. Because cilia lining the nose and lungs cannot beat effectively, mucus and trapped microbes linger, driving chronic sinus trouble, ear infections, and progressive lung damage. Many patients also have “situs inversus,” where the heart and other organs end up flipped left‑to‑right, a sign that cilia failed to guide the embryo’s early body plan. Doctors have known for some time that faults in dozens of different genes can cause PCD. ODAD1 is one of these genes and helps anchor the molecular motors that power each cilium’s whip‑like motion. But scientists still did not fully understand how ODAD1 defects play out inside human airway tissue.
Tracking a Faulty Gene in Patients’ Cells
The researchers studied nine individuals from seven Han Chinese families who showed classic signs of PCD: neonatal breathing problems, life‑long wet cough, frequent lung infections, and unusually low levels of nitric oxide in exhaled air, a clinical hallmark of the disease. Genetic testing revealed four harmful versions of the ODAD1 gene, including one never seen before. All of the faulty versions severely reduced or eliminated the normal ODAD1 protein in nasal airway cells. When the team filmed cilia from patient samples at high speed, they saw that the usual smooth, wave‑like beating was replaced by weak, disorganized flickering. Laboratory cultures grown from these cells, which recreate a thin, ciliated airway lining at an air–liquid interface, showed the same sluggish, uncoordinated motion.
Inside the Broken Micromachines
To see what was physically wrong, the scientists turned to powerful imaging techniques. Standard electron microscopy and cutting‑edge cryo‑electron microscopy revealed that the outer motor units and their docking sites were completely missing from patients’ cilia. In some cases, other key structures in the ciliary core were misplaced or malformed as well. These defects explain why the cilia cannot generate enough force to move mucus. Yet the damage went beyond the cilia themselves. The airway surface in patient‑derived cultures hosted far fewer multiciliated cells, and the remaining ones were oddly spaced and enlarged, with their cilia pointing in mixed directions. Surprisingly, the number of internal “basal bodies” per cell—the seeds from which cilia sprout—was normal, suggesting that the problem lay in how these cells were organized at the tissue surface rather than in how many cilia they tried to build.
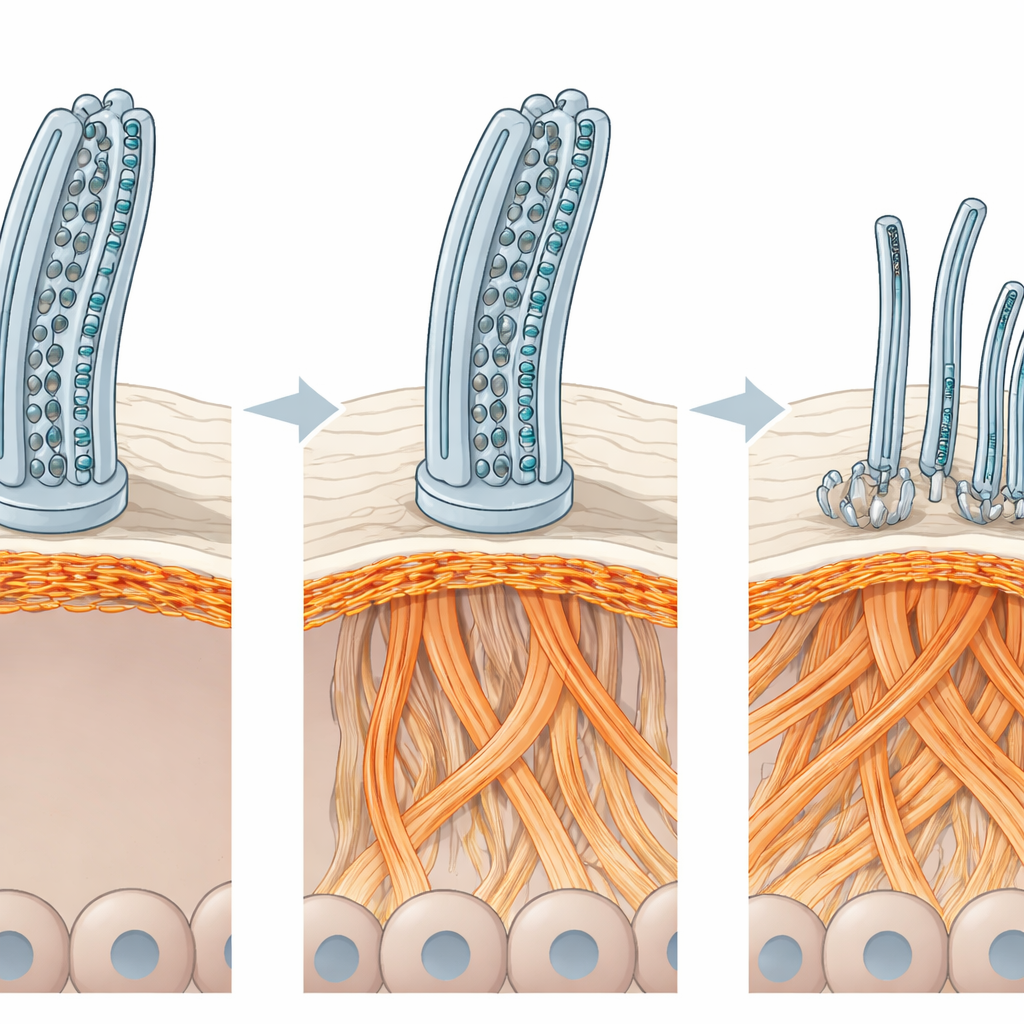
The Cell’s Inner Scaffold Gets Rewired
To pinpoint what was disrupting this organization, the team measured thousands of proteins in the patient‑derived airway cultures. While many cilia‑related proteins were reduced, several proteins linked to actin—the versatile fibers that form much of a cell’s internal scaffold—were more abundant. Imaging of actin filaments confirmed a dramatic reshaping of this scaffold: thickened actin bundles at the top of multiciliated cells, compressed networks at cell borders, and clumped patches deeper in the tissue. These changes were not unique to the patients’ genetic backgrounds; engineering an ODAD1 knockout in healthy cells produced the same actin rewiring and loss of multiciliated cells. When the researchers gently disrupted actin assembly with a small‑molecule drug, the number of multiciliated cells and their surface layout partially recovered, and cilia became more numerous and better organized—though still unable to beat normally without ODAD1’s motor‑docking role.
Restoring the Missing Part and Looking Ahead
Finally, the investigators tested whether replacing ODAD1 could revive ciliary motion. They grew “apical‑out” airway organoids—miniature, hollow spheres of airway tissue with cilia facing outward—from patient cells and used a lentiviral vector to deliver a working copy of ODAD1. The introduced protein correctly settled into cilia, restored the missing motor docking sites, and brought ciliary beating back close to normal speed and pattern. Together, these results show that ODAD1 loss harms the airways in two ways: it directly disables the cilia’s motor system and indirectly scrambles the actin scaffold that shapes the ciliated surface. For patients, this dual insight suggests a two‑pronged therapeutic future—gene therapies to fix the primary motor defect and safer actin‑modulating strategies to help rebuild a healthy carpet of cilia that can once again keep the lungs clear.
Citation: Huo, C., Luo, T., Yang, S. et al. Loss-of-function variants in ODAD1 disrupt ODA docking and induce actin cytoskeletal remodeling in primary ciliary dyskinesia. Cell Discov 12, 25 (2026). https://doi.org/10.1038/s41421-026-00875-8
Keywords: primary ciliary dyskinesia, ODAD1, motile cilia, actin cytoskeleton, gene therapy