Clear Sky Science · en
ATGL sensitizes hepatocellular carcinoma cells to genotoxic drugs by modulating p53 acetylation/phosphorylation status
Turning Fat Breakdown into a Cancer Weakness
Standard chemotherapy for liver cancer often fails because tumor cells are remarkably good at surviving DNA damage. This study explores an unexpected ally inside those very cancer cells: an enzyme that breaks down stored fat. By boosting this enzyme, called ATGL, the researchers found they could push liver tumor cells to stop repairing their DNA injuries and instead self‑destruct. The work uncovers a hidden link between how cancer cells handle fat and how they respond to powerful DNA‑damaging drugs, suggesting new ways to make existing treatments work better.
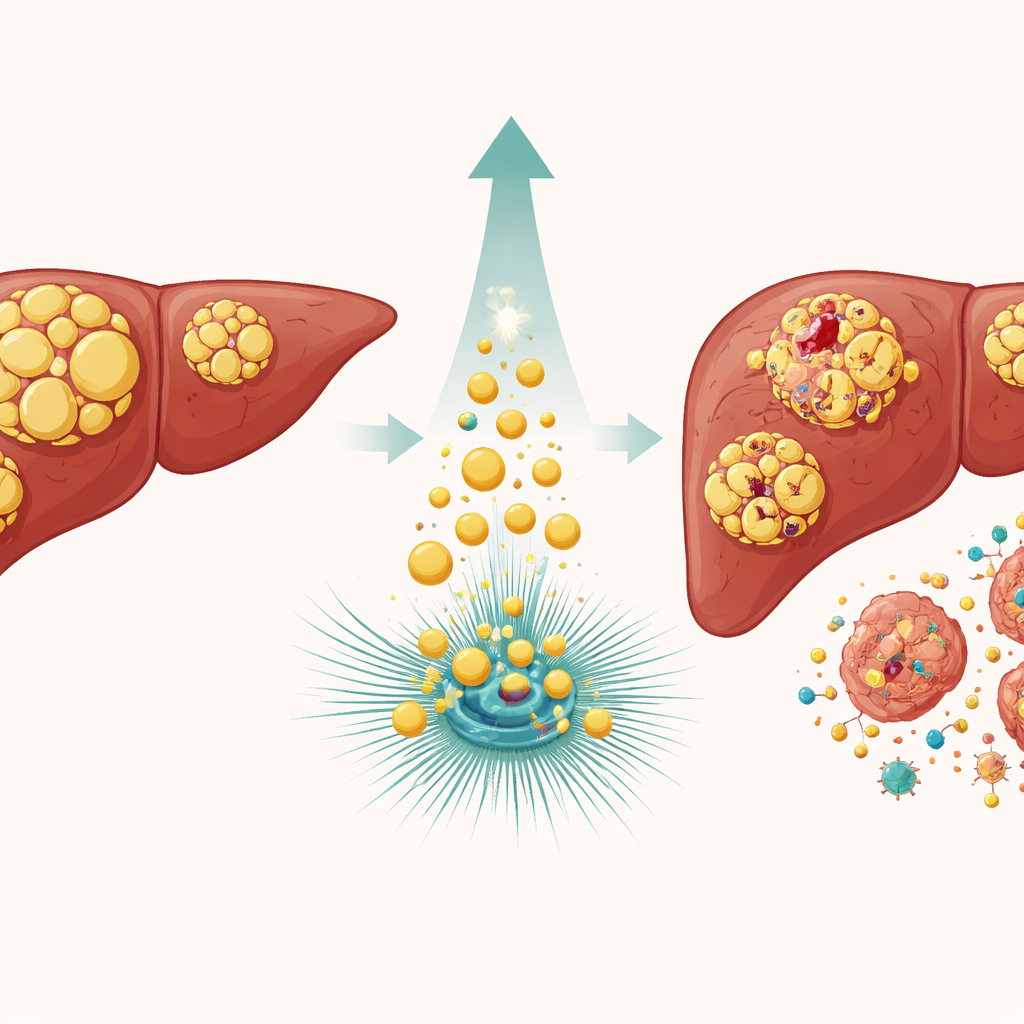
Why Liver Tumors Resist Harsh Drugs
Liver cancer, particularly hepatocellular carcinoma, is one of the most common and deadly tumor types worldwide. Many patients receive drugs that damage DNA, such as etoposide and doxorubicin, in hopes of forcing cancer cells into a lethal crisis. Yet these cells often escape by pausing their growth and activating repair systems controlled by a guardian protein known as p53. If the damage can be fixed, the cells resume dividing; if not, p53 can also trigger programmed cell death. The central puzzle is what tips p53 toward repair versus self‑destruction, and why some tumors remain so stubbornly resistant to therapy.
A Fat‑Cutting Enzyme Tips the Balance
The team focused on ATGL, an enzyme that trims stored fats in tiny cellular tanks called lipid droplets. In liver cancers, ATGL levels are typically lower than in healthy tissue, and earlier work hinted that forcing cells to make more ATGL slowed tumor growth. Here, the researchers engineered liver cancer cell lines to either overproduce ATGL or dial it down, then exposed them to DNA‑damaging drugs. Cells with extra ATGL accumulated far more signs of broken DNA, while cells with reduced ATGL showed less damage. Blocking ATGL’s cutting ability with a specific inhibitor, or expressing a mutant form that cannot work, erased this heightened sensitivity, proving that the enzyme’s fat‑breaking activity itself is key.
Rewiring the Cell’s Decision: Repair or Die
Diving deeper, the scientists examined p53, which acts like a molecular traffic cop after DNA injury. p53’s behavior is steered by small chemical tags added at specific positions. In ATGL‑rich cells, genotoxic drugs caused p53 to gain more of one type of tag (acetyl groups) and comparatively fewer of another (phosphate groups). This shift favored activation of genes that promote cell death, such as Puma, while dampening genes like p21 and GADD45 that normally pause the cell cycle and support DNA repair. As a result, even after the drug was washed away, ATGL‑high cells failed to clear markers of DNA damage and proceeded toward apoptosis instead of recovery.
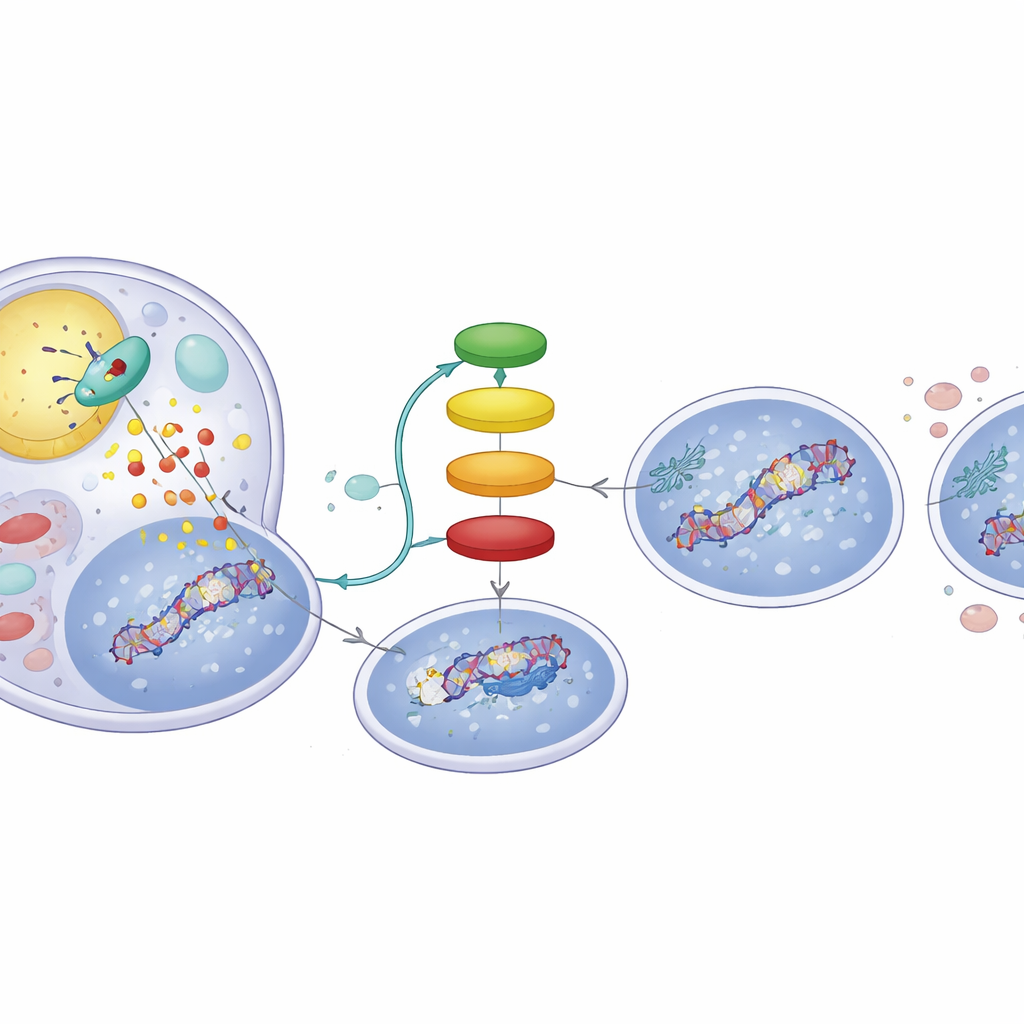
A Fat‑Driven Signal Chain Inside Tumor Cells
How does cutting up fat change p53’s tags? The breakdown products of ATGL’s action are free fatty acids that can act as messengers. The study shows that these fatty acids activate a nuclear receptor called PPARα, which in turn boosts the activity of p300, a protein that places acetyl tags on p53. When the researchers used a PPARα‑activating compound, they reproduced the ATGL‑high behavior: increased DNA damage signals and a p53 profile biased toward apoptosis. Conversely, blocking p300 wiped out the ATGL‑induced changes in p53 and reduced DNA damage, underscoring that an ATGL → PPARα → p300 chain is central to this switch. Analyses of hundreds of human liver tumors from public datasets echoed this link, revealing that tumors with higher ATGL expression also tend to show stronger PPARα and p300 signatures and expression of p53‑controlled genes.
What This Could Mean for Future Treatments
In plain terms, the study reveals that when liver cancer cells are encouraged to burn stored fat through ATGL, they become less inclined to repair chemotherapy‑induced DNA damage and more likely to undergo orderly cell death. This suggests two practical possibilities: measuring ATGL levels could help predict which patients’ tumors will respond better to genotoxic drugs, and boosting ATGL activity or its downstream PPARα pathway might be used alongside existing chemotherapies to overcome resistance. While further tests in animals and patients are needed, the work highlights a striking message: in liver cancer, making tumor cells "leaner" at the microscopic level may also make them more vulnerable to life‑saving drugs.
Citation: Castelli, S., De Cristofaro, A., Desideri, E. et al. ATGL sensitizes hepatocellular carcinoma cells to genotoxic drugs by modulating p53 acetylation/phosphorylation status. Cell Death Discov. 12, 164 (2026). https://doi.org/10.1038/s41420-026-03048-4
Keywords: hepatocellular carcinoma, ATGL, DNA damage response, p53 signaling, lipid metabolism in cancer