Clear Sky Science · en
Protein phosphatase 2A methylation state impacts α-synucleinopathy in mouse models
Why this matters for brain health
Parkinson’s disease and related disorders slowly rob people of movement, memory, and independence. A major culprit is a brain protein called alpha-synuclein that can misfold, clump, and damage nerve cells. This study asks a hopeful question: instead of attacking the protein directly, can we tune the brain’s own cleaning machinery to keep alpha-synuclein from becoming toxic?
A tale of a sticky protein
In Parkinson’s disease and Dementia with Lewy Bodies, twisted clumps of alpha-synuclein build up inside nerve cells, forming the classic “Lewy bodies.” A particular chemical tag on this protein, added at one spot called serine 129, is strongly linked to its most harmful form. When this tag is abundant, alpha-synuclein is more likely to form rigid fibrils and aggregates. The brain normally balances such tags using enzymes that add them and others that remove them. Because many enzymes can add the tag, blocking just one of them is unlikely to work. Instead, the authors focused on the main enzyme family that removes the tag, called protein phosphatase 2A, or PP2A, which acts as a molecular eraser for this dangerous modification.
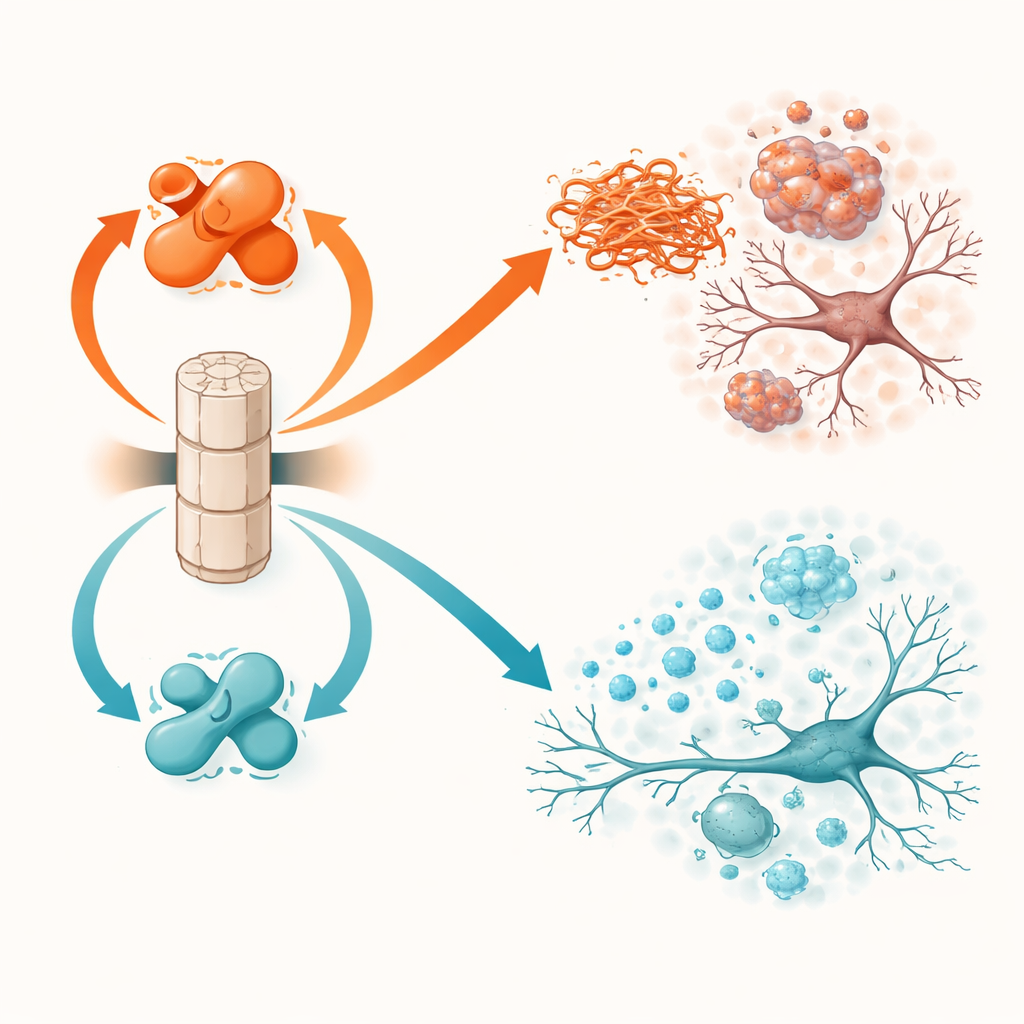
The brain’s eraser and its two switches
PP2A does not work at full strength by default. Its activity depends on a small chemical mark, called methylation, on one of its subunits. Two other proteins control this switch: LCMT-1 adds the mark and turns PP2A toward a more active, protective form, while PME-1 removes it and pushes PP2A toward a less active, harmful state. Earlier work in human brain tissue showed that in Parkinson’s and Lewy body dementia, LCMT-1 tends to be lower and PME-1 higher, leaving PP2A underpowered. The current study directly tests what happens when these switches are pushed deliberately in either direction in living mice that develop alpha-synuclein problems.
Testing the balance in living brains
The researchers used two complementary mouse models. In one, mice were engineered to produce human alpha-synuclein throughout the brain, gradually developing protein clumps and movement and memory problems with age. These animals were further altered to overproduce either PME-1 (the PP2A “off” switch) or LCMT-1 (the PP2A “on” switch) in forebrain neurons. In the second model, the team injected preformed alpha-synuclein fibrils into the striatum, a deep brain region involved in movement. These fibrils act as seeds, recruiting normal alpha-synuclein and spreading pathology over months in otherwise normal or enzyme-altered mice. In both models, the scientists measured protein buildup, nerve cell health, brain inflammation, and behavior.
When the eraser falters, damage spreads
Mice that overproduced PME-1, and thus had less active PP2A, fared worse. In alpha-synuclein transgenic animals, boosting PME-1 led to more heavily tagged and aggregated alpha-synuclein in the cortex and hippocampus, greater loss of nerve cell structure, weaker neuronal activity signals, and stronger activation of immune cells in the brain. These changes translated into poorer performance on movement tests and tasks of learning and memory. In the fibril-injection model, PME-1 overexpression allowed toxic alpha-synuclein assemblies to accumulate and spread more extensively, especially into the dopamine-producing nerve cells of the substantia nigra, a key region lost in Parkinson’s disease. These mice showed more severe loss of dopamine fibers, more intense inflammation, and greater motor and nesting deficits.
Turning the eraser back on
The opposite manipulation, overproducing LCMT-1 to keep PP2A strongly methylated and active, had broadly protective effects. In alpha-synuclein transgenic mice, LCMT-1 reduced the burden of tagged, aggregated protein to near-normal levels and preserved both the structure and activity of neurons. Inflammatory markers were lower, and animals performed closer to healthy controls on balance and memory tests. In the fibril-seeding model, LCMT-1 limited both the local buildup and the long-range spread of toxic alpha-synuclein, spared dopamine neurons from degeneration, reduced microglial activation, and blunted the decline in motor coordination and nesting behavior. Across experiments, shifting PP2A toward its active, methylated state consistently translated molecular benefits into functional protection.
What this could mean for future treatments
For non-specialists, the lesson is straightforward: the brain has a built-in eraser that can strip a harmful tag off alpha-synuclein and keep it from turning into dangerous clumps. When this eraser is weakened, damage, inflammation, and symptoms all get worse; when it is strengthened, nerve cells are shielded. The study provides direct evidence in living animals that the methylation state of PP2A is a master control for alpha-synuclein toxicity and its consequences. This points to a new therapeutic strategy: rather than chasing every harmful form of the protein, drugs could be designed to nudge PP2A and its regulators LCMT-1 and PME-1 toward a more protective setting. Such approaches will require careful safety testing, but they hold promise for slowing or preventing Parkinson’s disease and related conditions by restoring the brain’s own ability to keep alpha-synuclein in check.
Citation: Maddila, S., Hassanzadeh, K., Liu, J. et al. Protein phosphatase 2A methylation state impacts α-synucleinopathy in mouse models. Cell Death Discov. 12, 172 (2026). https://doi.org/10.1038/s41420-026-03045-7
Keywords: Parkinson’s disease, alpha-synuclein, protein phosphatase 2A, neurodegeneration, brain inflammation