Clear Sky Science · en
Mitochondrial bioenergetics-SASP crosstalk determines senolytic efficacy in therapy-induced senescence
Why zapping “zombie” cancer cells is so tricky
Many modern cancer drugs don’t kill every tumor cell outright. Instead, some cells enter a limbo-like state called senescence: they stop dividing but stay alive, a bit like “zombie” cells. These therapy-induced senescent cells can be helpful at first, but if they linger they may fuel relapse, resistance, and side effects. Scientists are therefore testing senolytic drugs designed to selectively kill senescent cells. This paper asks a deceptively simple question: why do some senescent cancer cells die when exposed to senolytics, while others stubbornly survive?
Power plants that remember their past
At the heart of the study are mitochondria, the tiny power plants inside cells. The authors examined whether the way mitochondria burn different fuels—such as sugars, fats, and amino acids—affects how sensitive senescent cancer cells are to senolytic drugs that target a survival protein called BCL‑xL. Using a high-throughput assay (MitoPlate S‑1), they functionally “fingerprinted” mitochondrial activity in several cancer cell lines before and after senescence was triggered by different treatments (DNA-damaging drugs, mitotic blockers, oxidative stress, or cell-cycle inhibitors). They found that therapy-induced senescence did not produce a single, uniform mitochondrial state. Instead, each drug left a distinct “bioenergetic imprint,” changing how broadly and intensely mitochondria could tap into various energy sources. Crucially, this flexibility was layered on top of a pre-existing baseline: the parental cancer cells’ original mitochondrial configuration set an upper limit—the “ceiling”—on how strong any later senolytic response could be.
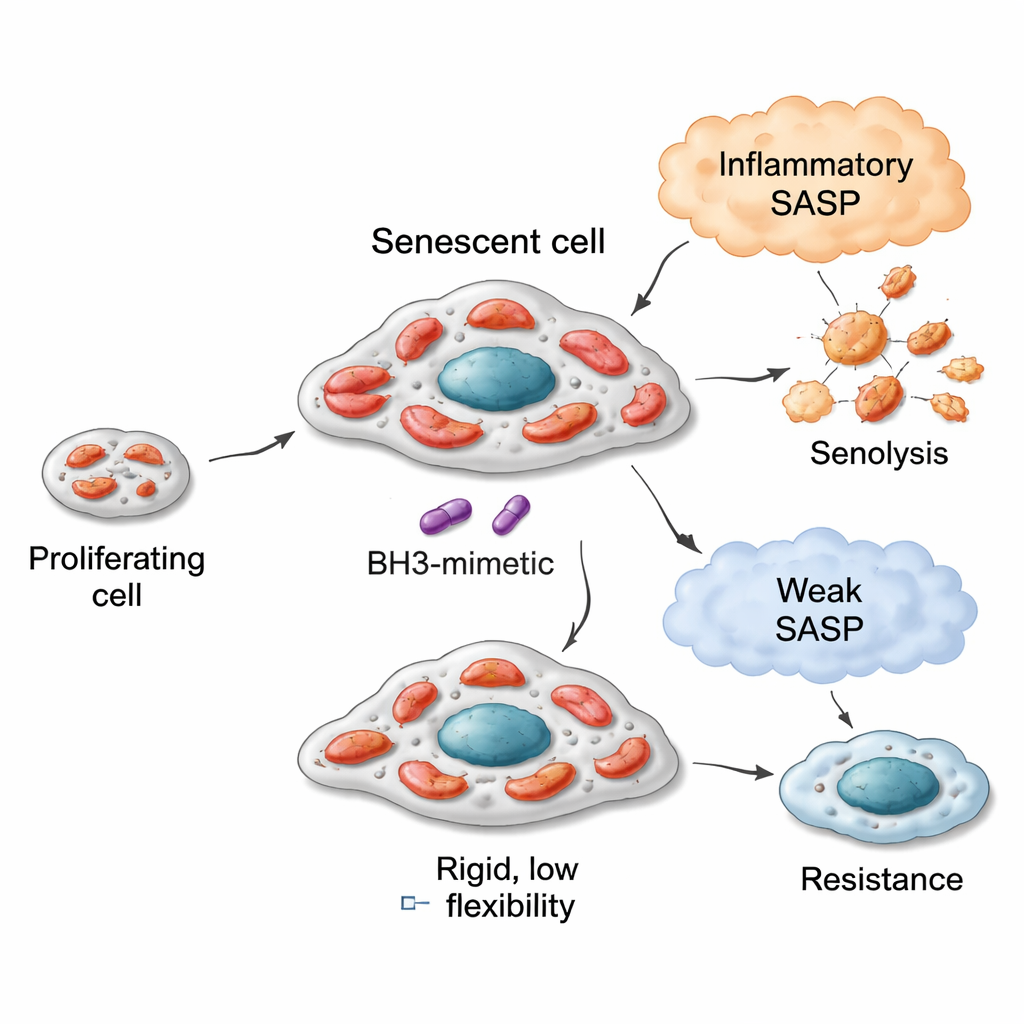
Flexible engines, different fuels, and senolytic sensitivity
In lung, breast, and colon cancer models, senescent cells with more flexible mitochondria—able to oxidize a wider array of fuels—tended to be more vulnerable to BCL‑xL–targeting senolytics like navitoclax (ABT‑263) and A1331852. For example, lung cancer cells driven into senescence by the drug bleomycin developed mitochondria that vigorously used many substrates, especially those linked to fatty acid breakdown and certain amino acid pathways. These cells were highly sensitive to senolytics. In contrast, cells pushed into senescence by a CDK4/6 inhibitor (palbociclib) showed a narrower metabolic repertoire and responded poorly to the same senolytic agents. Yet this relationship had limits: breast cancer cells could also become metabolically flexible after senescence, but because their starting mitochondria were less “primed” for apoptosis, their maximum senolytic response was modest. Colon cancer cells with defective apoptotic machinery remained resistant regardless of how their metabolism shifted. A single measure—how well cells oxidized the fuel succinate at baseline—served as a simple indicator of this inherited mitochondrial capacity.
When metabolism talks to inflammation
Senescent cells are notorious for the SASP, a cocktail of secreted inflammatory and growth factors that can influence surrounding tissues. The team probed how mitochondrial metabolism connects to this secretory behavior using cells engineered with a reporter for miR‑146a, a microRNA switched on by the master inflammatory regulator NF‑κB. They found that while overall SASP profiles were largely dictated by the cell type, only certain senescent states activated this NF‑κB/miR‑146a axis. Those were the same states that responded well to BCL‑xL senolytics. Importantly, these “inflammatory SASP–positive” senescent cells also showed enhanced use of fatty acid oxidation and a transcriptional upshift in genes that shuttle long-chain fats into mitochondria. Blocking fatty acid entry with the drug etomoxir dampened miR‑146a activation without fully reversing senescence, suggesting that mitochondrial fuel choice helps license an inflammatory, senolytic-permissive SASP.
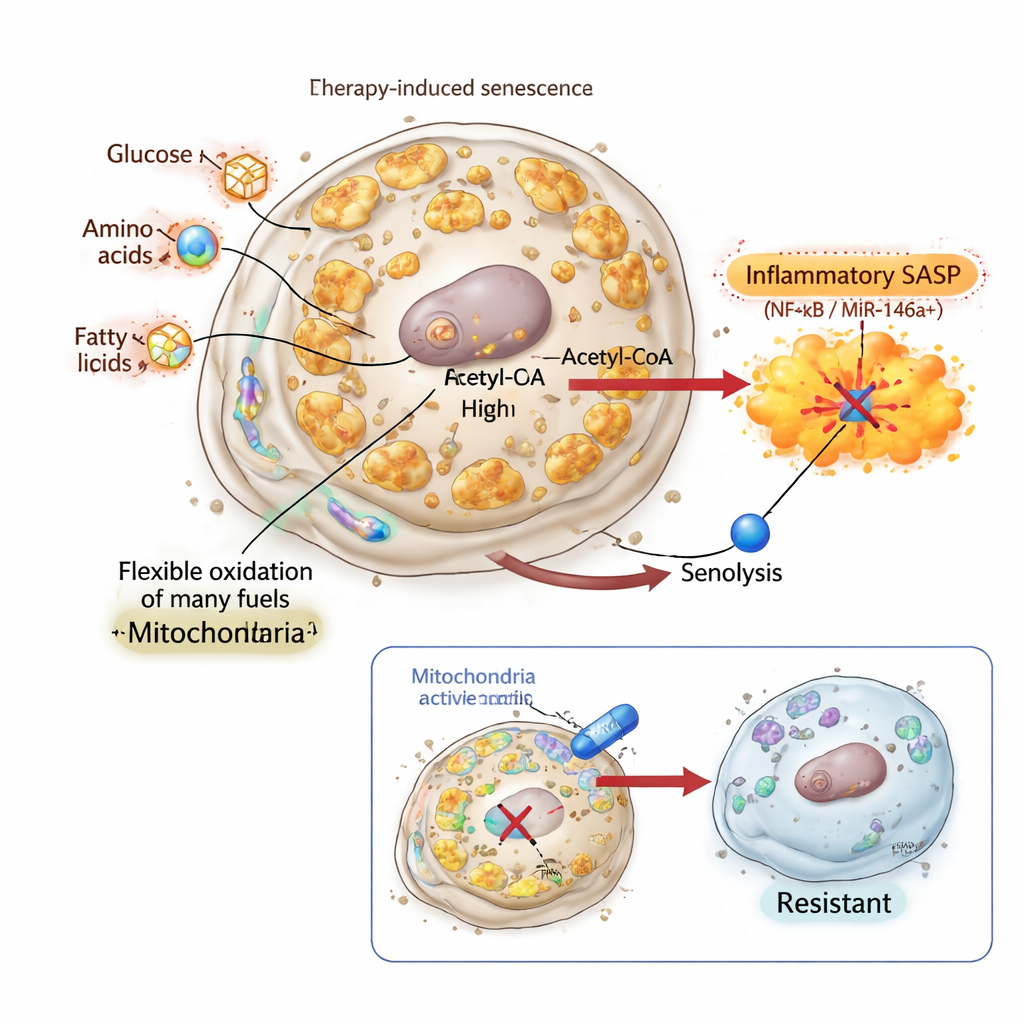
Silencing the signal and creating unkillable senescent cells
To test whether the inflammatory SASP is truly required for senolytic killing, the researchers turned to inflachromene, a compound that binds chromatin proteins HMGB1 and HMGB2 and blocks their role in activating SASP genes. In lung and breast cancer cells, inflachromene induced a textbook senescent phenotype: cells became large, stopped dividing, and accumulated senescence markers. Their mitochondrial mass and bioenergetic activity rose substantially, and their fuel usage was clearly remodeled. Yet their SASP was blunted and the miR‑146a reporter remained largely silent. Strikingly, these SASP-null senescent cells were completely resistant to BCL‑xL–targeting senolytics, despite having bioenergetically active, reprogrammed mitochondria and reduced expression of the classic anti-apoptotic gene BCL2. This showed that mitochondrial changes alone are not enough; without a mitochondria-driven inflammatory output, the senolytic “second punch” fails.
What this means for future cancer treatments
For a lay reader, the study’s conclusion is that killing therapy-induced “zombie” cancer cells is governed by a layered circuit. First, the original health and wiring of a tumor cell’s mitochondria set how far senolytic drugs can ever go. Second, the particular treatment that causes senescence can push mitochondrial metabolism toward more or less flexibility, moving cells closer to or farther from the brink of apoptosis. Third—and most decisively—senolytic drugs only work well if metabolic rewiring successfully engages an inflammatory SASP program that talks back to the nucleus. Without that inflammatory cross-talk, senescent cells can become a dead-end, drug-resistant state. Practically, this suggests that future therapies might be optimized by functionally testing both mitochondrial flexibility and SASP inflammation in tumors, then choosing senescence-inducing drugs and senolytics in combinations that ensure the “zombie” cells are not just frozen in place, but primed for removal.
Citation: Llop-Hernández, À., Verdura, S., López, J. et al. Mitochondrial bioenergetics-SASP crosstalk determines senolytic efficacy in therapy-induced senescence. Cell Death Discov. 12, 103 (2026). https://doi.org/10.1038/s41420-026-02967-6
Keywords: cellular senescence, mitochondria, senolytics, cancer metabolism, inflammatory SASP