Clear Sky Science · en
Caspase-activation powers anti-Desmoglein 3-induced acantholysis in human epidermis
When the Body Turns on Its Own Skin Glue
Pemphigus vulgaris is a rare but dangerous disease in which the body’s immune system attacks the “glue” that holds skin cells together, causing painful blisters and open sores. This study looks under the microscope to answer a key question: do these blisters form only because antibodies block that glue, or do additional cell-death signals help rip skin cells apart? Understanding this could lead to more targeted and gentler treatments for patients.
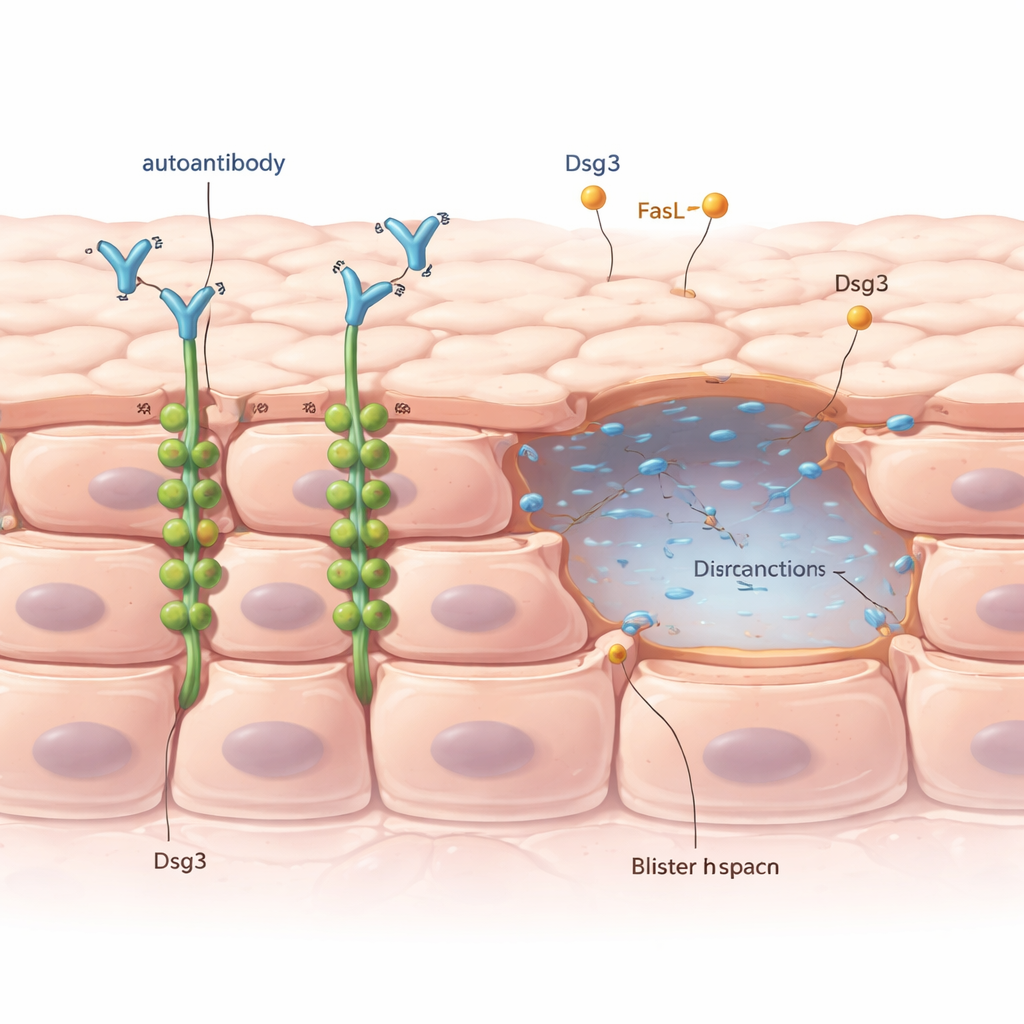
How Skin Cells Normally Stay Stuck Together
The outer layer of our skin is made of tightly packed cells connected by tiny structures called desmosomes, which act like rivets between neighboring cells. A key component of these rivets is a protein called desmoglein 3, or Dsg3 for short. In pemphigus vulgaris, patients produce antibodies that mistakenly latch onto Dsg3. Doctors have long known that this antibody attack leads to a process called acantholysis, where skin cells lose contact and blisters appear. But puzzlingly, blisters only show up in certain places and at certain times, even though the harmful antibodies are spread throughout the skin.
Antibodies Alone Can Start the Damage
To see what the antibodies themselves do, the researchers used pieces of healthy human skin and a lab-grown skin cell line. They exposed them to a well-studied anti-Dsg3 antibody. They found that this antibody could weaken cell–cell adhesion and trigger acantholysis even without switching on the usual cell-death machinery inside the cells. The key event was Dsg3 being pulled away from the cell surface into the cell’s interior, often packaged into small vesicles. This removal of Dsg3 from the “rivets” between cells made the desmosomes shorter and less effective, allowing cells to start separating.
When Cell-Death Signals Join In, Things Get Worse
The team then asked whether a known cell-death signal in pemphigus, a molecule called Fas ligand (FasL), changes this picture. FasL can activate enzymes called caspases, especially caspase-8, which normally drive cells toward programmed death. In patient skin samples and in their skin model, the researchers saw active caspase-8 in areas with damage but no classic signs of dying cells. When they added small, non-lethal amounts of FasL together with the anti-Dsg3 antibody, blisters formed more quickly and more severely, particularly in the deeper layers of the epidermis. In cell culture, FasL alone did not weaken adhesion, but in combination with the antibody it greatly increased cell break-up—and this boost disappeared when caspase-8 was blocked.
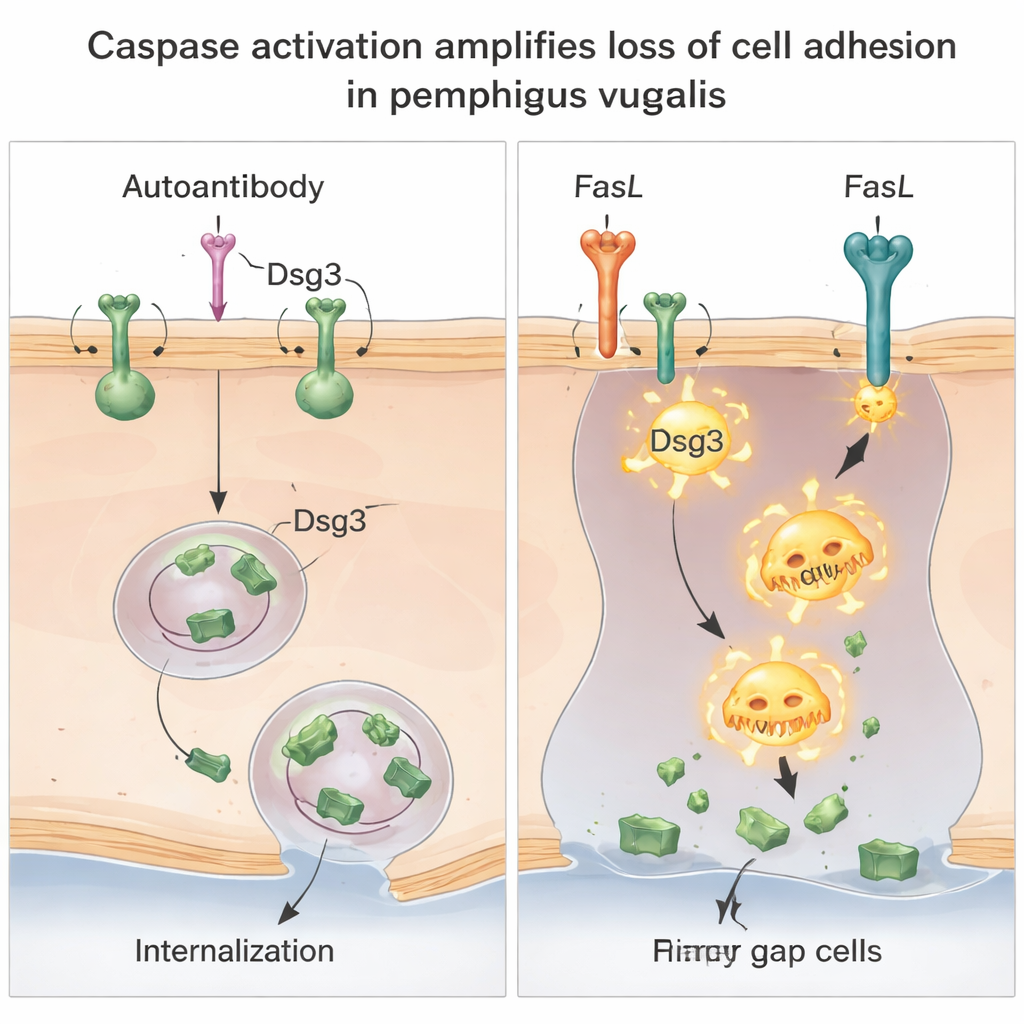
Two Ways to Break the Same Bond
Digging deeper, the scientists showed that there are actually two distinct but cooperating ways that Dsg3 is lost. First, the antibody promotes the internalization of intact Dsg3–antibody complexes from the cell surface into specialized membrane compartments, reducing the amount of Dsg3 available at cell junctions. This step did not depend on caspases and was not stopped by blocking common protein-degradation routes, implying a specific reshuffling of Dsg3 inside the cell. Second, when FasL is present, caspase-8 becomes active and cuts Dsg3 into smaller fragments, especially in the more loosely attached parts of the membrane. This caspase-driven cutting further lowers the amount of full-length, functional Dsg3 and magnifies the loss of adhesion triggered by the antibodies.
What This Means for Patients and Future Treatments
Taken together, the results suggest that in pemphigus vulgaris, antibodies against Dsg3 start the blistering process by stripping Dsg3 from the cell surface, while FasL-driven caspase activation acts like an amplifier that makes the damage much worse by cleaving the remaining Dsg3. Importantly, much of this happens before the skin cells actually die. For patients, this means that effective therapies may not only need to reduce harmful antibodies but also block FasL or caspases to prevent blisters from forming or spreading. This dual mechanism may help explain why the disease can look and behave so differently from one person—or one body site—to another, and points toward new, more precise ways to keep the skin’s natural glue intact.
Citation: Schmidt, M.F., Feoktistova, M.A., Panayotova-Dimitrova, D. et al. Caspase-activation powers anti-Desmoglein 3-induced acantholysis in human epidermis. Cell Death Discov. 12, 102 (2026). https://doi.org/10.1038/s41420-026-02963-w
Keywords: pemphigus vulgaris, autoimmune blistering, desmoglein 3, caspase-8, Fas ligand