Clear Sky Science · en
Modulating metabolic signatures to mitigate cabozantinib resistance in FLT3-ITD acute myeloid leukemia cell models
Why this matters for cancer treatment
Many modern cancer drugs are designed to home in on a single faulty protein in tumor cells. These targeted drugs can trigger dramatic remissions, but cancers often find ways to adapt and grow back. This paper looks at how a type of blood cancer, acute myeloid leukemia (AML), becomes resistant to one such targeted drug, cabozantinib, and how re-wiring the cancer cells’ energy use might help doctors outsmart that resistance.
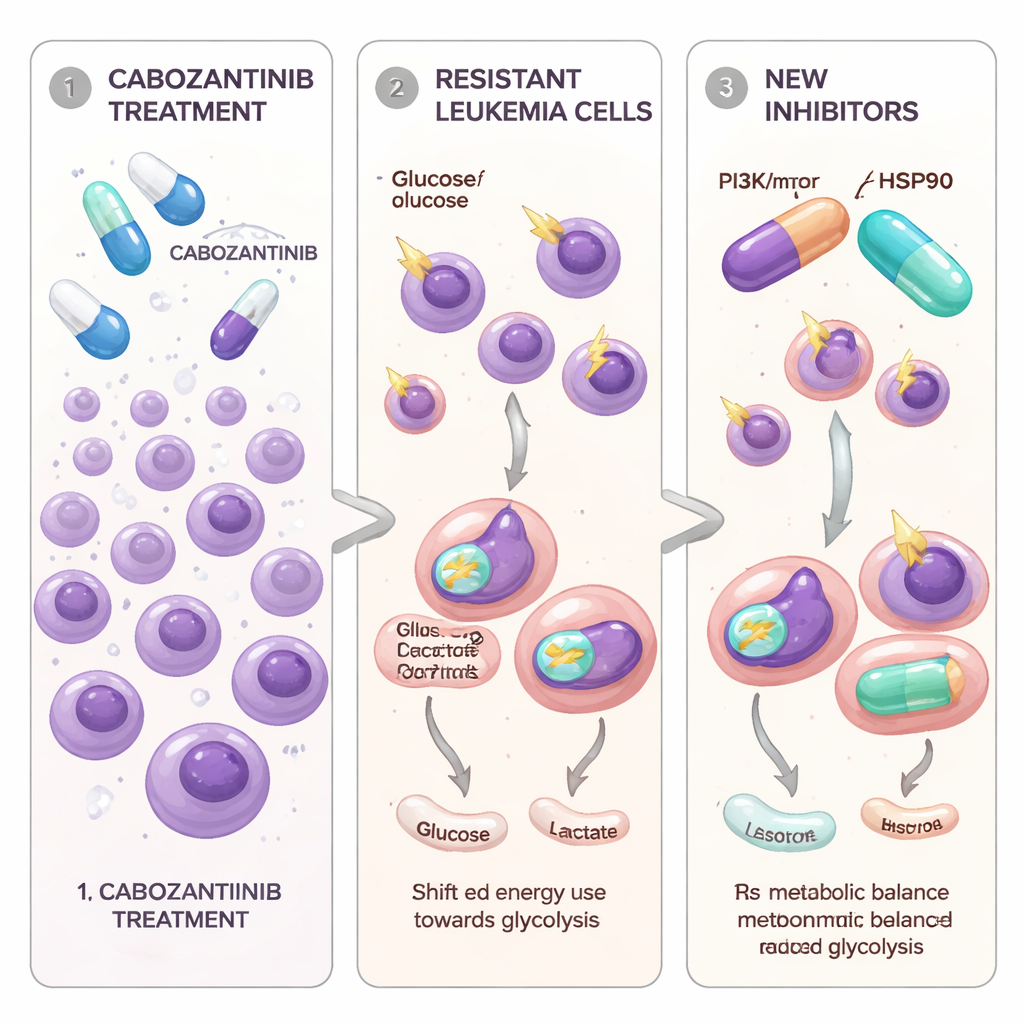
Leukemia cells that learn to dodge a targeted drug
The researchers focused on AML cells that carry a mutation in a growth‑signal switch called FLT3-ITD, which is known to drive especially aggressive disease. Cabozantinib, a pill already used for several solid tumors, can strongly block FLT3‑driven leukemia cells in the lab. To model what happens in patients over time, the team gradually exposed two FLT3‑mutant AML cell lines to rising doses of cabozantinib until some cells survived and began to grow again. These new cell populations, called Molm13‑XR and MV4‑11‑XR, could tolerate cabozantinib concentrations many times higher than their original “parent” cells. They also became less sensitive to two other approved FLT3‑targeting drugs, sorafenib and quizartinib, while remaining vulnerable to a different inhibitor, gilteritinib.
Genetic tweaks that help cancer survive
Looking under the hood, the scientists found that these drug‑resistant leukemia cells carried new changes in their FLT3 gene. Both resistant lines had acquired the same point mutation, called D835Y, in a crucial region of FLT3’s kinase domain, a known hotspot for resistance to several drugs. One line, MV4‑11‑XR, also gained an unusual 1.3‑kilobase deletion that removed an entire exon from FLT3, erasing part of the domain important for drug binding. These changes appeared to have been selected during long‑term cabozantinib exposure: mutant versions of FLT3 became much more common in the resistant cells than in the starting population. At the same time, key signaling pathways downstream of FLT3—such as ERK, STAT5, and AKT—were more strongly switched on, supporting faster growth and greater colony formation in the resistant cells.
Cancer cells switch fuel systems
The team then asked whether resistance was linked not only to genetics, but also to how the cells fuel themselves. Using RNA sequencing and dedicated metabolic tests, they found a consistent pattern: cabozantinib‑resistant cells relied much more heavily on glycolysis—the rapid breakdown of glucose in the cell fluid—even when oxygen was plentiful. These cells took up more glucose, produced more lactate, showed higher activity of a key enzyme called GAPDH, and turned up the expression of multiple glycolysis‑related genes. In contrast, the cells’ mitochondria, the structures that support more efficient energy production, were less active and less abundant. Measurements of oxygen use revealed that both basic and peak mitochondrial respiration were reduced, and reactive oxygen species inside the cells were elevated, pointing to stressed, underperforming mitochondria.
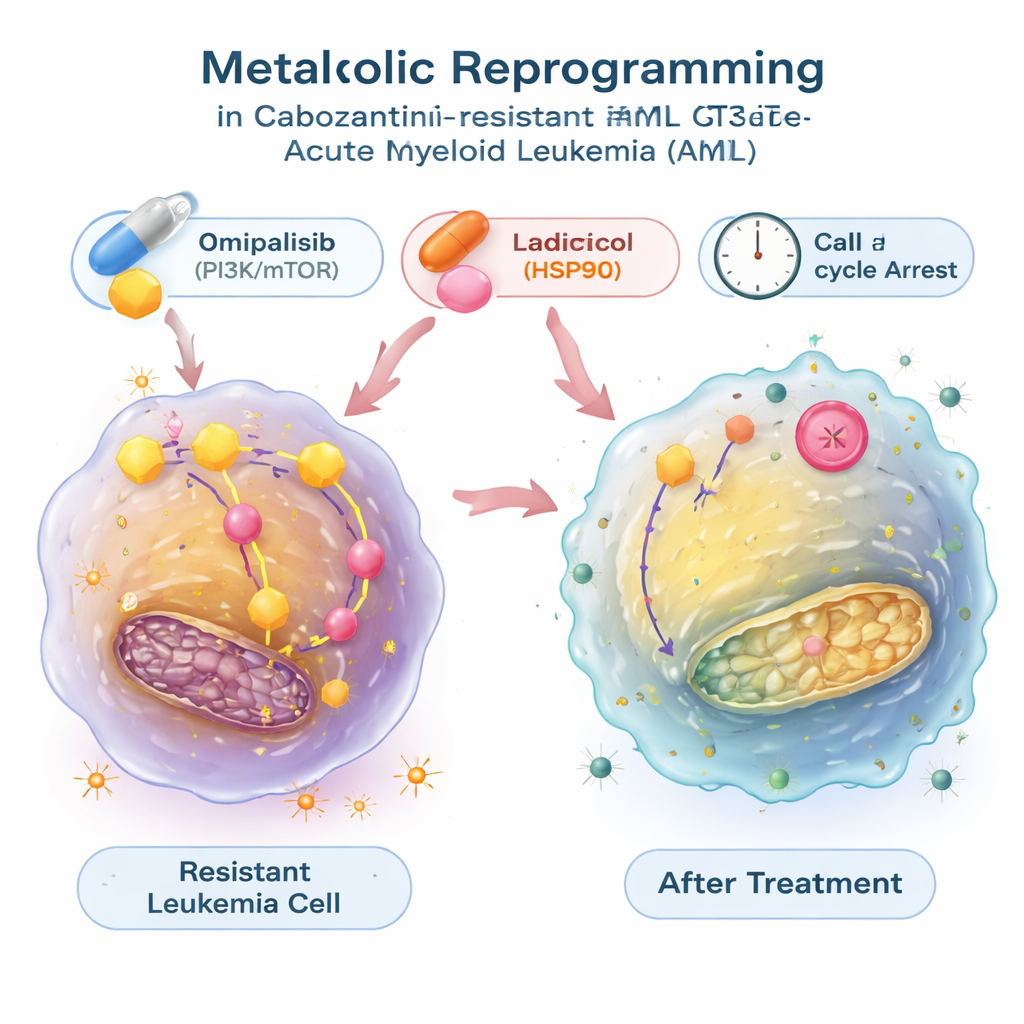
Finding drugs that flip the metabolic switch back
To see if this energy shift could be reversed, the researchers used a large public database that connects gene‑expression patterns with the effects of thousands of compounds. They searched for drugs predicted to counteract the metabolic signature of the resistant leukemia cells and homed in on two: radicicol, which blocks a protein chaperone called HSP90, and omipalisib, which inhibits the PI3K/mTOR signaling pathway that controls growth and metabolism. In lab tests, both molecules not only slowed the growth of resistant cells but also reduced their overactive glycolysis, normalizing glucose uptake and lactate release and dialing down glycolysis‑related genes. These compounds pushed leukemia cells into a resting phase of the cell cycle, and in the case of radicicol, also triggered substantial programmed cell death. When combined with cabozantinib, omipalisib—and, in one model, radicicol—worked synergistically, making the drug‑resistant cells easier to kill.
What this means for future leukemia therapies
For non‑specialists, the message is that leukemia cells can escape a targeted drug not only by mutating its direct target, but also by changing how they make and use energy. The study shows that cabozantinib‑resistant AML cells adopt a “sugar‑burning” strategy while letting their mitochondria languish. By hitting the pathways that support this rewired metabolism—through drugs like omipalisib or HSP90 inhibitors—it may be possible to restore sensitivity to cabozantinib and similar treatments. Although these findings come from cell models rather than patients, they suggest that pairing targeted cancer drugs with metabolism‑modulating agents could be a promising way to delay or overcome resistance in FLT3‑mutant AML.
Citation: Fu, YH., Ng, K.M., Tseng, CY. et al. Modulating metabolic signatures to mitigate cabozantinib resistance in FLT3-ITD acute myeloid leukemia cell models. Cell Death Discov. 12, 98 (2026). https://doi.org/10.1038/s41420-026-02957-8
Keywords: acute myeloid leukemia, drug resistance, FLT3 mutation, cancer metabolism, cabozantinib