Clear Sky Science · en
DNA polymerase kappa stabilized by Ptbp2 interacts with MRE11 and promotes genomic instability in leukemia
How Leukemia Cells Keep Broken DNA and Still Survive
Our DNA is constantly under attack, yet healthy cells are usually very good at finding and fixing damage. In leukemia, however, some cells learn to live with broken, unstable DNA—and even turn that instability into a survival advantage. This study uncovers a molecular “team-up” between a splicing protein (Ptbp2), a special DNA-copying enzyme (DNA polymerase kappa, or Polk), and a damage-sensing factor (MRE11) that helps leukemia cells repair just enough damage to stay alive, while accumulating the genetic chaos that drives cancer progression.
A Hidden Helper in Leukemia Cells
The researchers focused on chronic myeloid leukemia (CML), a blood cancer usually driven by the BCR::ABL1 fusion gene. While modern drugs that block BCR::ABL1 work well in early disease, many patients who reach the aggressive “blast crisis” phase respond poorly. Previous work showed that Ptbp2, a protein that binds RNA and influences how messages are processed, is boosted by BCR::ABL1 and acts like an oncogene in CML. Here, the team discovered that Ptbp2 latches onto the tail end (3′ UTR) of the Polk messenger RNA and protects it from being degraded. As a result, leukemia cells make more Polk protein whenever Ptbp2 levels are high.
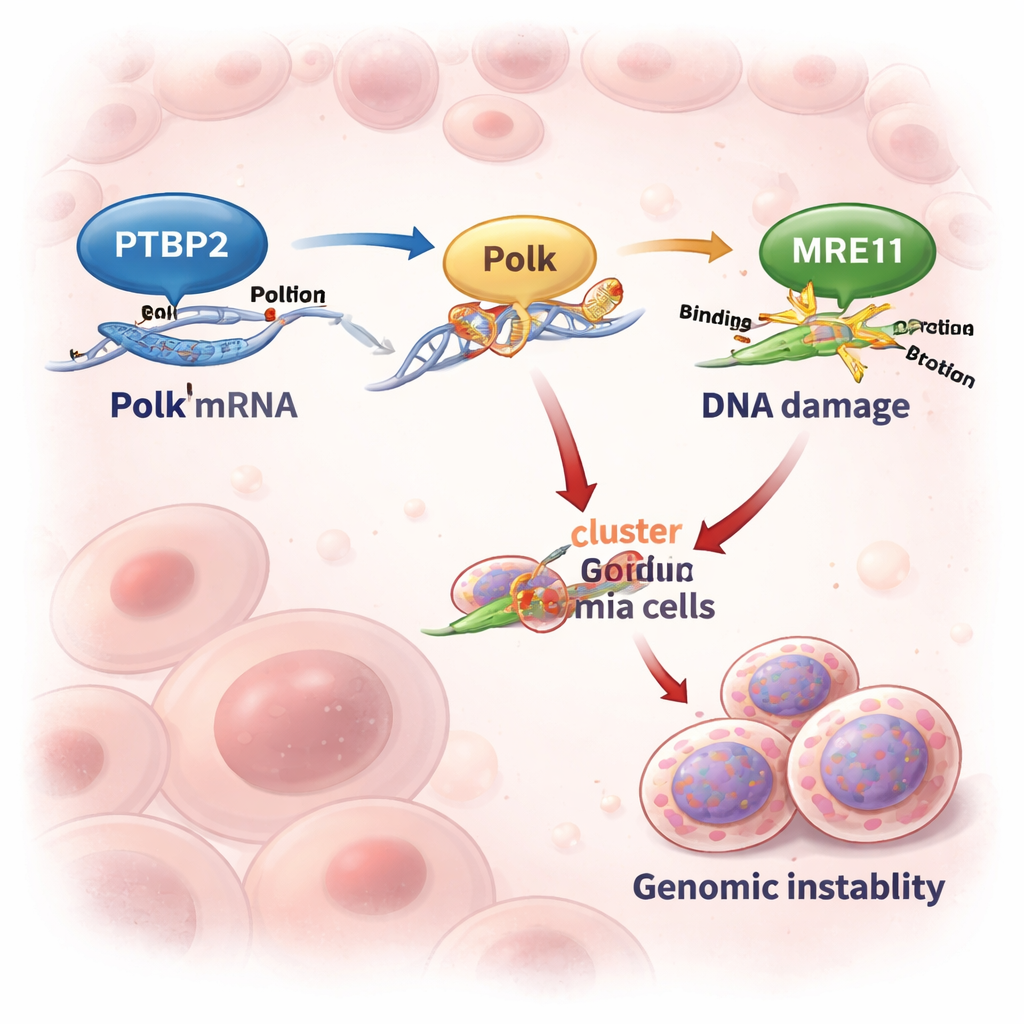
Turning On an Error-Prone DNA Copymachine
Polk is a “backup” DNA polymerase that can copy over damaged DNA when the usual replication machinery stalls. This ability can rescue stressed cells—but it comes at a price, because Polk is error-prone and can introduce mutations. In cell lines and patient samples from advanced CML, Ptbp2 and Polk levels rose and fell together. When scientists knocked out Ptbp2 in leukemia cells, Polk levels dropped sharply, and the Polk RNA decayed almost twice as fast. Re-introducing Polk into Ptbp2-deficient cells restored their behavior, showing that Ptbp2’s main role here is to keep Polk abundant and active.
Repairing Damage—But Not Perfectly
To see how this duo affects DNA repair, the researchers treated cells with hydroxyurea, a drug that stalls DNA replication and is often used in CML patients. Cells lacking Ptbp2 suffered much more DNA damage, visible as long “comet tails” and bright γH2AX foci—hallmarks of broken chromosomes. These damaged cells were more likely to die. In contrast, cells with high Ptbp2 and Polk better tolerated the drug, repaired damage more efficiently, and survived, even though their repair was sloppy. Overexpression of Polk in Ptbp2-knockout cells rescued this sensitivity, confirming that the Ptbp2–Polk partnership helps leukemia cells get through replication stress and avoid apoptosis.
A DNA Damage Network That Favors Instability
The story does not end with Polk. The team showed that Polk physically interacts with MRE11, a key member of the MRN complex that senses DNA breaks and activates the ATM–CHK2 damage-response pathway. When Ptbp2 was removed, Polk dropped, MRE11 levels and activity declined, and ATM–CHK2 signaling weakened. Putting Polk back restored MRE11 and its activation. Detailed DNA-fiber experiments revealed that Ptbp2 and Polk help protect stalled replication forks from being chewed back, largely through MRE11. Blocking MRE11 with a drug undermined this fork protection and increased DNA damage. Paradoxically, cells with active Ptbp2–Polk–MRE11 signaling accumulated more chromosomal abnormalities, such as sister chromatid exchanges, breaks, gaps, multipolar spindles, and multinucleated giant cells—classic signs of genomic instability that can fuel more aggressive cancer.
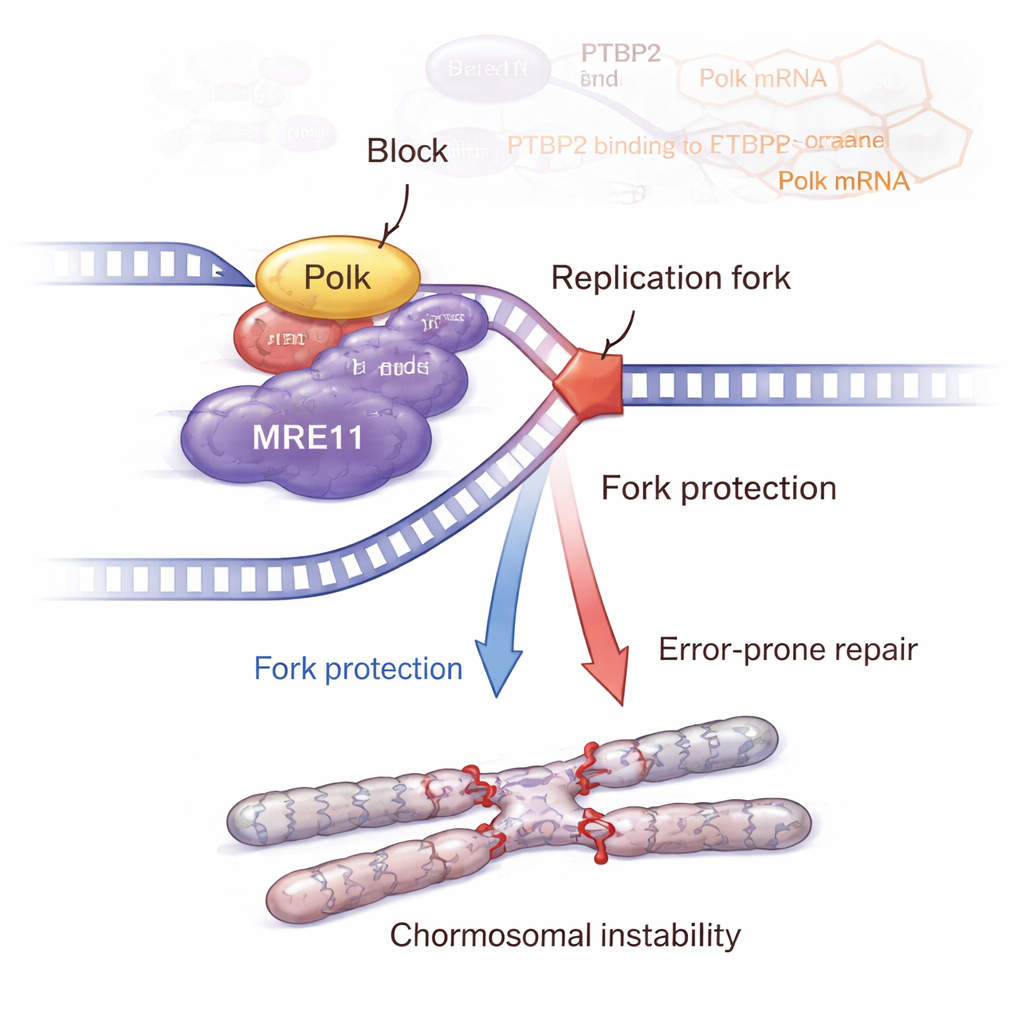
From Mice to Possible New Treatments
In mouse models, leukemia cells with intact Ptbp2 produced larger, more abnormal tumors than cells lacking Ptbp2. Tissues from these mice showed higher levels of Ptbp2, Polk, the proliferation marker Ki-67, and distorted cell division structures. In a separate CML-like mouse model driven by BCR::ABL1, adding extra Ptbp2 boosted Polk and increased the number of atypical dividing cells and invasive leukemia clusters in spleen and liver, pointing to faster disease progression. Together, these findings suggest that the Ptbp2–Polk–MRE11–ATM–CHK2 axis allows leukemia cells to survive intense DNA stress while steadily building up harmful mutations.
Why This Matters for Patients
For a layperson, the key message is that some leukemia cells escape control by carefully walking a tightrope: they fix their DNA just enough to stay alive, but not so well that they avoid mutations. Ptbp2 stabilizes Polk, which then teams up with MRE11 to protect stressed DNA and keep damage signals running—yet this repair is imperfect and promotes genetic chaos. Because advanced CML and other cancers seem to depend on this fragile balance, targeting Ptbp2 or its control over Polk could tip cells away from survival and toward self-destruction, offering a promising new angle for therapies, especially in the hard-to-treat blast crisis stage.
Citation: Lama, S., Barik, B., IS, S. et al. DNA polymerase kappa stabilized by Ptbp2 interacts with MRE11 and promotes genomic instability in leukemia. Cell Death Discov. 12, 96 (2026). https://doi.org/10.1038/s41420-026-02951-0
Keywords: chronic myeloid leukemia, genomic instability, DNA repair, DNA polymerase kappa, PTBP2