Clear Sky Science · en
Adipogenic transdifferentiation reprograms EMT-high PDAC cells into a post-mitotic adipocyte-like state and limits metastasis
Turning Aggressive Cancer Cells into Harmless Fat
Pancreatic cancer is one of the deadliest cancers, in large part because it spreads early and resists standard treatments. This study explores a strikingly different idea: instead of trying to poison or starve tumor cells, what if we could coax the most dangerous pancreatic cancer cells to transform into fat-like cells that no longer divide or travel through the body? The work describes how researchers pushed highly aggressive pancreatic cancer cells toward a stable, fat-cell–like state that slowed tumor growth and reduced spread in mice, hinting at a new way to control this devastating disease.
Why Pancreatic Cancer Is So Hard to Stop
Pancreatic ductal adenocarcinoma, the main form of pancreatic cancer, has a very poor outlook: only about one in eight patients is alive five years after diagnosis. Part of the problem is that many tumor cells in this cancer live in a shape-shifting state called EMT, which makes them more mobile, invasive, and resistant to drugs. Attempts to block the many signals that drive this state have brought limited benefit. At the same time, the pancreas and its cancers show a surprising tendency to accumulate fat cells, raising the question of whether this hidden flexibility in cell identity could be harnessed. The authors reasoned that if EMT-heavy pancreatic cancer cells are already primed to change, perhaps they could be redirected into a quiet, fat-like identity rather than an invasive one.
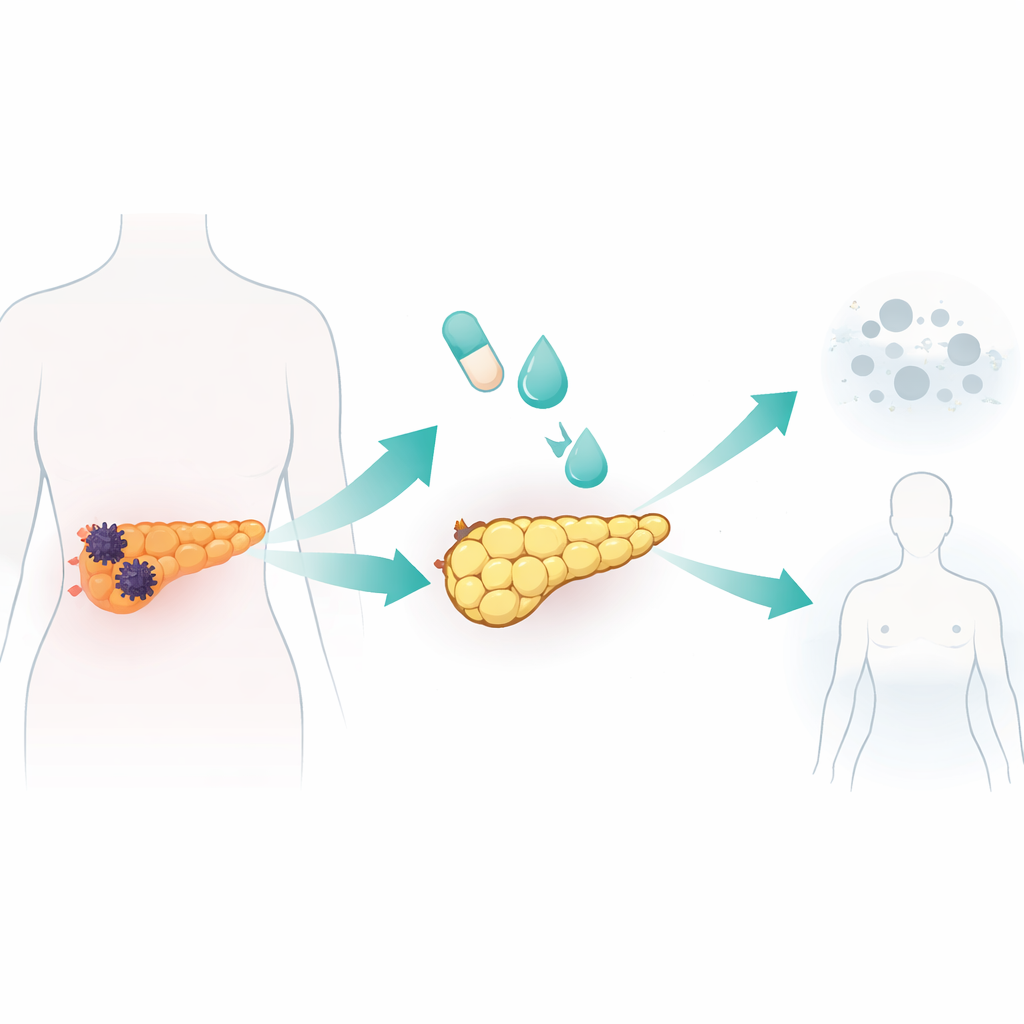
Reprogramming Cancer Cells into Fat-Like Cells in the Lab
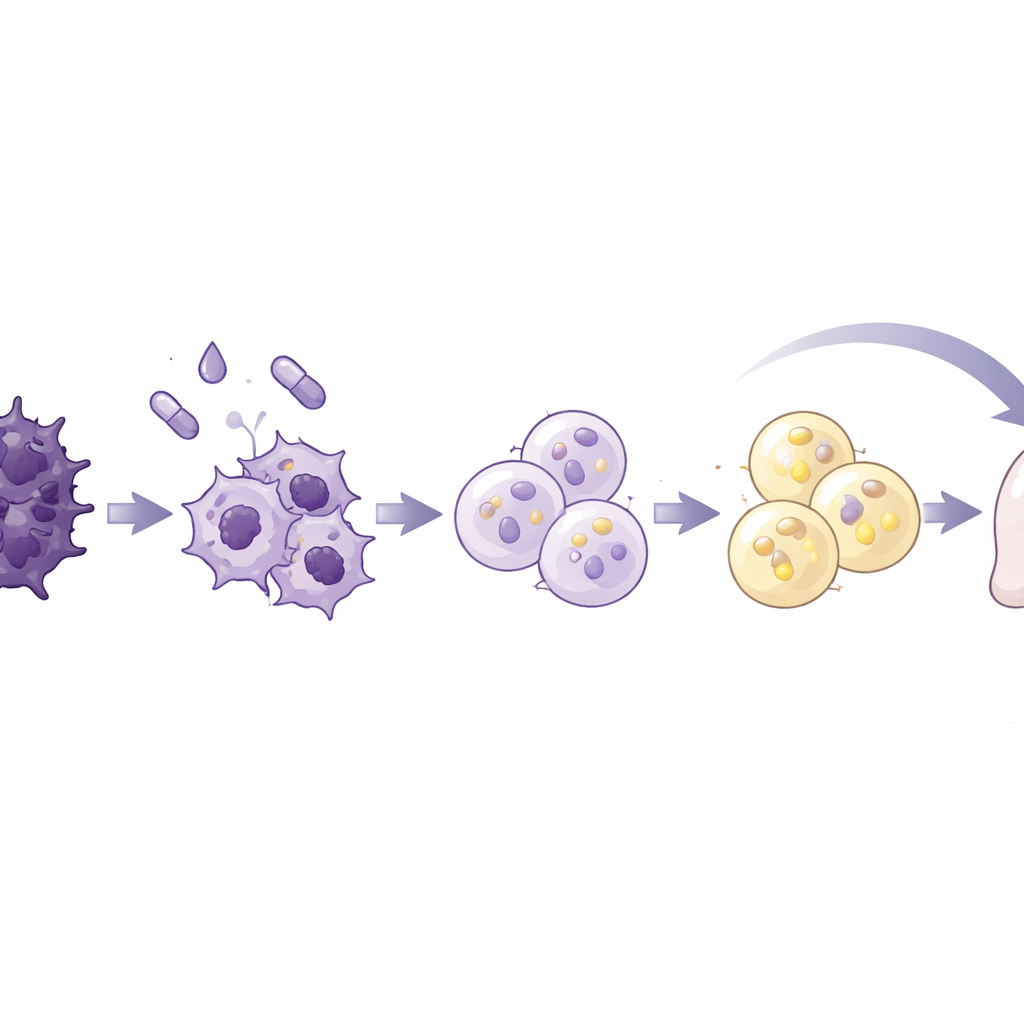
The team tested a standard fat-cell–forming cocktail, used in earlier fat biology and breast cancer studies, on seven human pancreatic cancer cell lines and one normal pancreatic cell line. The mixture combined insulin and a steroid with rosiglitazone, a drug that activates a master fat regulator, plus a signaling protein called BMP2 to unlock cell plasticity. One cancer line, called AsPC-1, was especially responsive. Over ten days, these cells became larger and rounder and filled with lipid droplets, hallmarks of fat cells. They switched on genes and proteins typical of mature fat cells and showed robust fat metabolism, including secreting adiponectin and breaking down stored fat on cue. Crucially, these converted cells stopped proliferating, stalled in the early phase of the cell cycle, and moved and invaded far less than untreated cancer cells.
Shutting Down the Cancer Program Deep Inside the Cell
To see what was happening at a molecular level, the researchers profiled the converted cells’ DNA packaging and gene activity. They found a broad tightening of chromatin, the DNA–protein complex that controls gene access, along with a global drop in gene expression, both features of nondividing cells. Genes that support EMT, invasion, and metastasis, including matrix-degrading enzymes and key EMT regulators, were strongly repressed, while fat-related genes were boosted. The overall gene signature shifted from a mesenchymal, highly mobile identity to one that closely resembled mature fat cells. Signals linked to cell growth and response to a major EMT driver, TGF-beta, were toned down, whereas pathways related to lipid handling and cell adhesion were enhanced. These changes suggest that the cells were not merely slowed, but fundamentally re-specified.
Putting the Strategy to the Test in Mice
The scientists next asked whether this forced fat-like conversion could help control tumors in living animals. They implanted human pancreatic cancer cells into the pancreas or spleen of mice to model primary tumors and liver metastases. Mice treated with rosiglitazone and BMP2 developed smaller pancreatic tumors and showed richer lipid droplets and fat-cell markers within the tumors, along with lower levels of EMT and invasion genes. In the metastasis model, treatment did not change early tumor seeding, but over time it slowed the expansion of liver tumor burden compared with untreated animals. Importantly, neighboring normal pancreatic tissue did not show obvious fat-cell conversion, and the fat-like state in tumors persisted for at least a month after the drugs were stopped, suggesting a degree of durability and specificity.
What This Could Mean for Future Cancer Care
This study supports a provocative concept: for highly plastic, EMT-rich pancreatic cancers, it may be possible to “convert instead of kill,” redirecting dangerous, wandering tumor cells into stable, nondividing fat-like cells that are less able to spread. While this is early-stage work in cells and mouse models, and not all pancreatic cancers are equally responsive, it opens a new therapeutic avenue that acts by changing cell identity rather than just blocking growth signals. In the future, such transdifferentiation approaches might be combined with targeted drugs or immunotherapies to hold pancreatic cancer in a quieter, more manageable state and reduce the risk of lethal metastasis.
Citation: Qian, Y., Yan, Z., Wang, J. et al. Adipogenic transdifferentiation reprograms EMT-high PDAC cells into a post-mitotic adipocyte-like state and limits metastasis. Cell Death Dis 17, 330 (2026). https://doi.org/10.1038/s41419-026-08613-4
Keywords: pancreatic cancer, cell plasticity, transdifferentiation, epithelial-mesenchymal transition, adipocyte-like cells