Clear Sky Science · en
Loss of PRKACB facilitates metastasis of diffuse-type gastric cancer through RhoA signaling activation
Why this study matters for stomach cancer
Diffuse-type gastric cancer is a particularly aggressive form of stomach cancer that often spreads widely inside the abdomen, making it hard to treat and deadly. This study digs into why these tumors are so prone to spreading, uncovering a molecular braking system that normally restrains cancer cells from wandering—and showing what happens when that brake fails. Understanding this hidden control switch could open new paths for predicting which patients are at highest risk and for developing drugs that slow or stop dangerous spread.
A dangerous form of stomach cancer
Not all stomach cancers behave the same. Intestinal-type tumors tend to form more structured masses, while diffuse-type gastric cancer (DGC) is made of scattered cells that readily slip away from the primary tumor. Patients with DGC have a higher risk of death than those with intestinal-type disease, in part because their tumors more easily seed new growths throughout the abdominal cavity. Earlier genetic studies had already linked DGC to recurring changes in a gene called RHOA, which influences how cells move and change shape. But it was unclear how these changes connected to the broader signaling networks that control whether cancer cells stay put or migrate.
Finding a missing brake in tumor samples
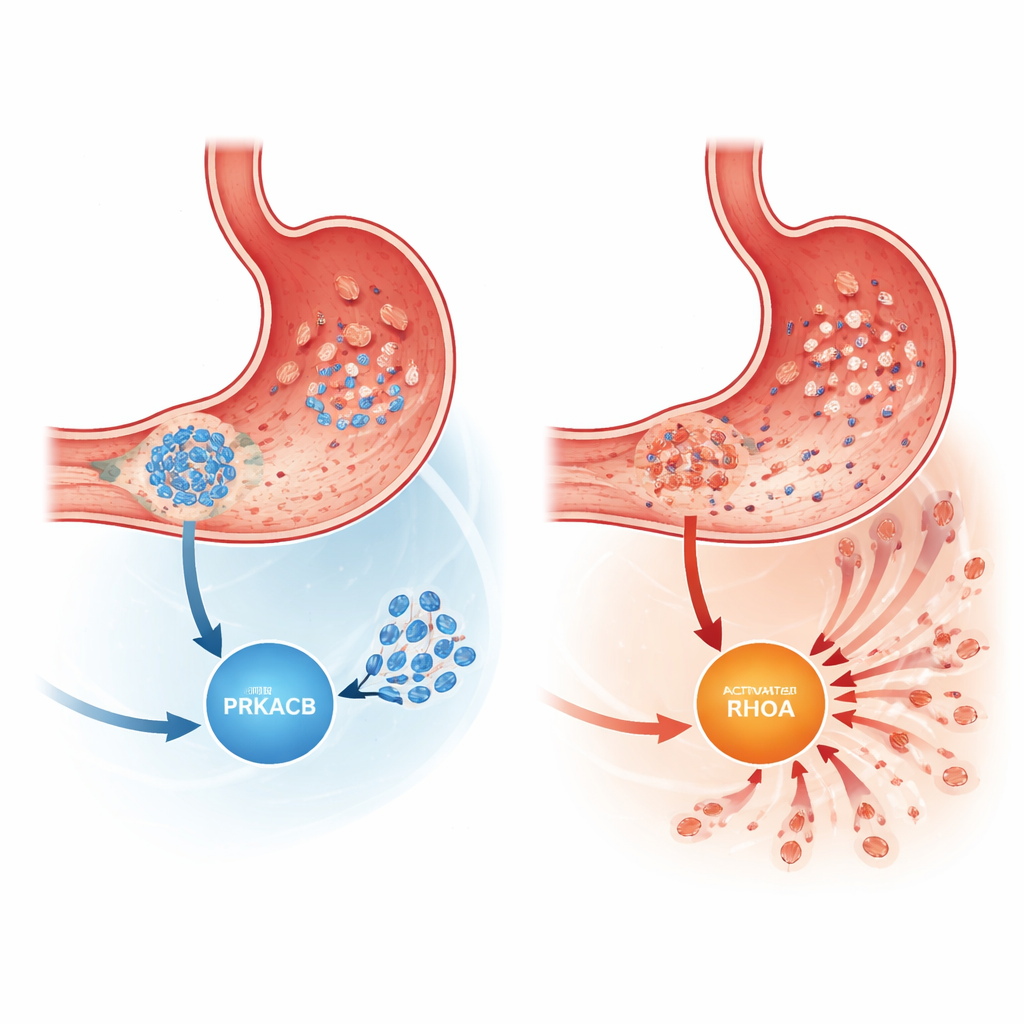
The researchers focused on a protein called PRKACB, a catalytic subunit of the well-known enzyme protein kinase A, which fine-tunes many cell behaviors by adding phosphate groups to other proteins. An earlier proteomics survey had hinted that PRKACB levels were unusually low in aggressive DGC. To test this, the team examined tumor samples from 376 patients, comparing diffuse-type and intestinal-type cancers and their nearby noncancerous tissue. They found that PRKACB levels were markedly reduced in diffuse-type tumors, especially in advanced stages, but not in intestinal-type cancers. Patients whose tumors had little PRKACB had significantly worse overall survival, even after accounting for other clinical factors, suggesting PRKACB functions as a tumor suppressor in this setting.
How low PRKACB fuels cell escape
To see what PRKACB does inside cancer cells, the team used cultured cell lines that model diffuse-type gastric cancer. When they artificially reduced PRKACB, the cells became more mobile and invasive in lab tests, more readily squeezing through barriers and forming finger-like projections called pseudopodia that help cells crawl. These cells also shifted from a more orderly, epithelial state toward a looser, mesenchymal-like state associated with metastasis, losing the adhesion molecule E‑cadherin that normally helps cells stick together. Conversely, boosting PRKACB levels made the cells less migratory and less invasive. Importantly, these changes did not alter how fast the cells multiplied, pointing to a specific role for PRKACB in enabling spread, not growth.
Zooming in on the RhoA signaling switch
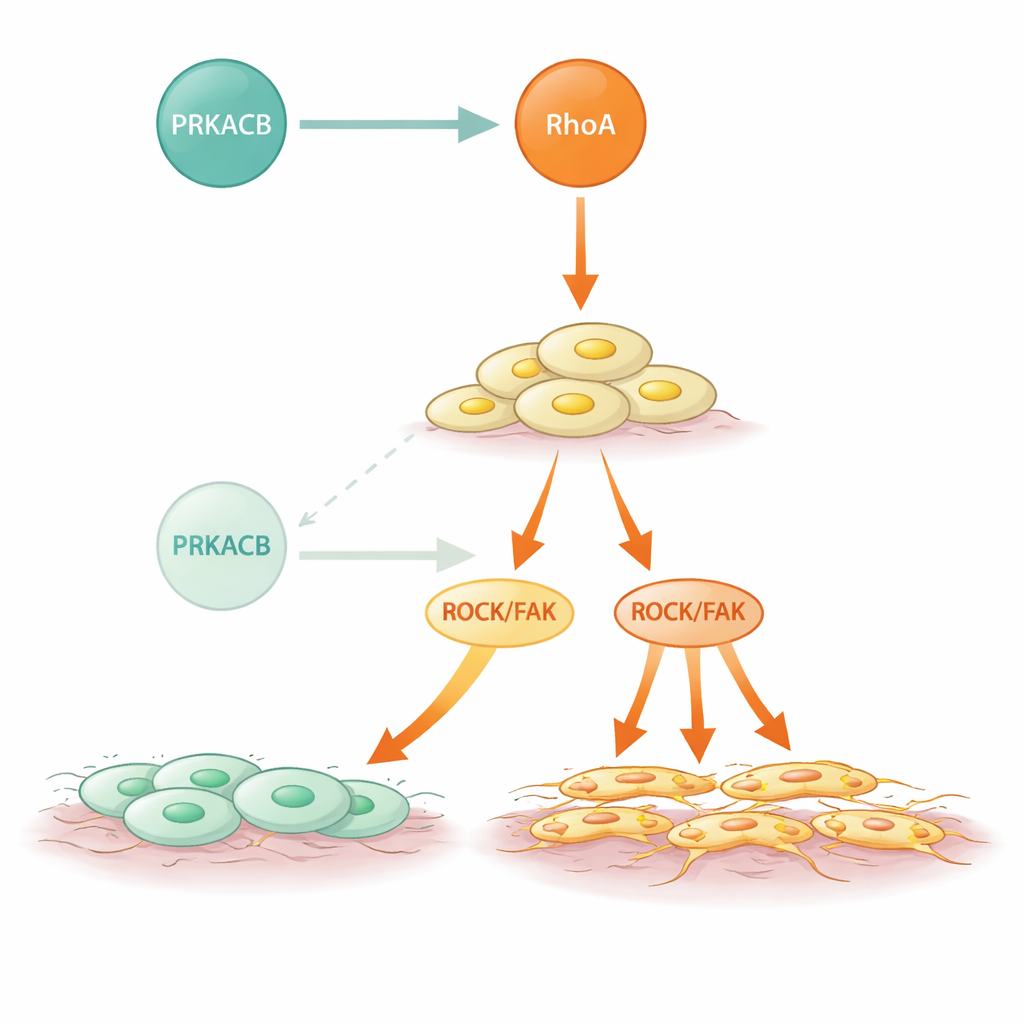
The researchers then asked how PRKACB exerts this anti-metastatic effect. Using protein–protein interaction methods and structural modeling, they showed that PRKACB directly binds to RhoA, a molecular switch that toggles between inactive and active forms to govern cell shape and movement. PRKACB adds a phosphate group at a specific position on RhoA (serine 188), which turns RhoA down and dampens the downstream ROCK and FAK pathways that drive cytoskeletal rearrangements and cell motility. When PRKACB was knocked down, RhoA was less phosphorylated, more active, and ROCK/FAK signaling intensified, leading to more aggressive cell movement. In mouse models where human-like tumors were implanted into the stomach wall, lowering PRKACB caused more and larger metastatic nodules in the abdomen and earlier onset of visible spread, again without changing how quickly established metastases grew.
Mutant signaling and a possible way to intervene
Diffuse-type gastric cancers often harbor RHOA mutations, and this study shows how some of those changes magnify the problem. Several frequent DGC-associated mutations in RhoA weakened or abolished its binding to PRKACB but did not impair RhoA’s ability to activate its downstream partners. As a result, these mutant forms escaped the restraining phosphorylation by PRKACB and showed heightened ROCK activity and stronger invasive behavior. Strikingly, when the researchers treated cells and mice with a RhoA-blocking compound, the extra metastasis caused by low PRKACB was largely reversed. This suggests that even in tumors where the natural brake is weak or missing, targeting the overactive RhoA pathway directly could curb spread.
What this means for patients and future therapies
In simple terms, this work identifies PRKACB as a key part of an internal “anti-migration” circuit in diffuse-type gastric cancer. When PRKACB levels fall, or when RhoA is mutated so that PRKACB can’t bind and modify it, RhoA signaling runs hotter, and cancer cells become more adept at breaking away and colonizing the abdominal cavity. Measuring PRKACB and RhoA status in tumors could help doctors gauge how likely a patient’s cancer is to metastasize and who might benefit most from drugs that inhibit the RhoA–ROCK–FAK pathway. While such treatments will require more development and clinical testing, the study maps a clear molecular route from a missing brake to deadly spread—and points to new ways of slowing that journey.
Citation: Sun, J., Zhao, J., Yang, X. et al. Loss of PRKACB facilitates metastasis of diffuse-type gastric cancer through RhoA signaling activation. Cell Death Dis 17, 281 (2026). https://doi.org/10.1038/s41419-026-08553-z
Keywords: diffuse-type gastric cancer, metastasis, PRKACB, RhoA signaling, ROCK FAK pathway