Clear Sky Science · en
PD-1 protects expanding human T cells from premature restimulation-induced cell death by modulating TCR and CD28 signaling
Why stopping friendly fire in the immune system matters
Our immune systems rely on armies of T cells that rapidly multiply to fight infections and cancer. But this explosive growth is dangerous: if too many T cells remain active for too long, they can damage healthy tissues or fuel autoimmune disease. This study explores how a well-known “brake” on T cells, a molecule called PD‑1, helps expanding human T cells avoid dying too soon from their own over-activation. Understanding this balance is crucial for safer cancer immunotherapies and for preventing harmful immune overreactions.
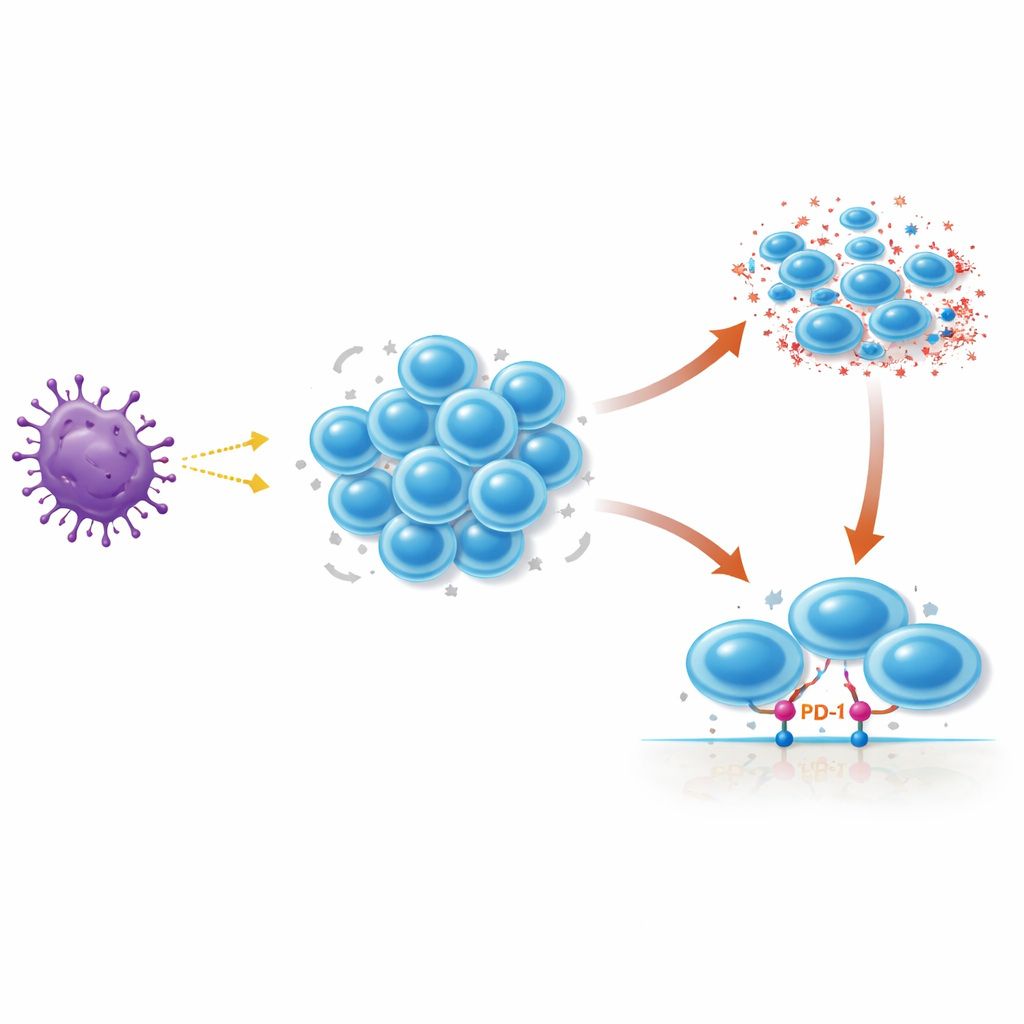
A built-in self-destruct that must be carefully timed
Once T cells recognize a threat, they divide into large clones of identical fighters. To prevent this response from running out of control, T cells carry a built-in self-destruct program called restimulation-induced cell death (RICD). When an already-activated T cell is strongly triggered again through its main sensor, the T cell receptor, RICD can be switched on to kill that cell. This helps shrink the army after a threat is cleared and avoids dangerous lymphoproliferative diseases where T cells accumulate excessively. However, early in a response, T cells need time to expand before this self-destruct pathway becomes dominant, and how that timing is controlled in humans has not been fully clear.
The surprising protective side of an immune brake
PD‑1 is famous as a target of “checkpoint” drugs used in cancer treatment, where blocking it can reawaken exhausted T cells inside tumors. Traditionally seen as a molecule that weakens T cell activity, PD‑1 is rapidly switched on when human T cells first encounter a stimulus and remains present at moderate levels during their early expansion. In this study, researchers isolated human CD4 and CD8 T cells from healthy donors, activated them in culture, and tracked PD‑1 and its partner PD‑L1 over time. They found that PD‑1 and PD‑L1 peak within a few days of activation—precisely when T cells are multiplying—and then decline as cells mature. When the team blocked PD‑1 or PD‑L1 during repeated stimulation, more T cells died, indicating that normal PD‑1 signaling actually shields these expanding cells from premature RICD.
How PD‑1 reshapes the conversation at the cell surface
To probe this protection more precisely, the scientists built artificial “proxy” antigen-presenting beads coated with activating signals (to mimic the T cell receptor and its main helper, CD28) and, in some cases, PD‑L1. When expanding T cells were restimulated with beads carrying PD‑L1, they consistently lost far fewer cells than with control beads, and standard markers of early and late-stage apoptosis dropped toward baseline. This survival boost depended on how much PD‑L1 was present and on having PD‑L1 positioned right next to the activating signals, echoing the tight organization of the natural immune synapse. Interestingly, the protective effect was strongest when CD28 was engaged together with the T cell receptor, suggesting that PD‑1 reins in both activating pathways at once. Without PD‑1, CD28 made cells more prone to RICD, but adding PD‑L1 erased this extra sensitivity.
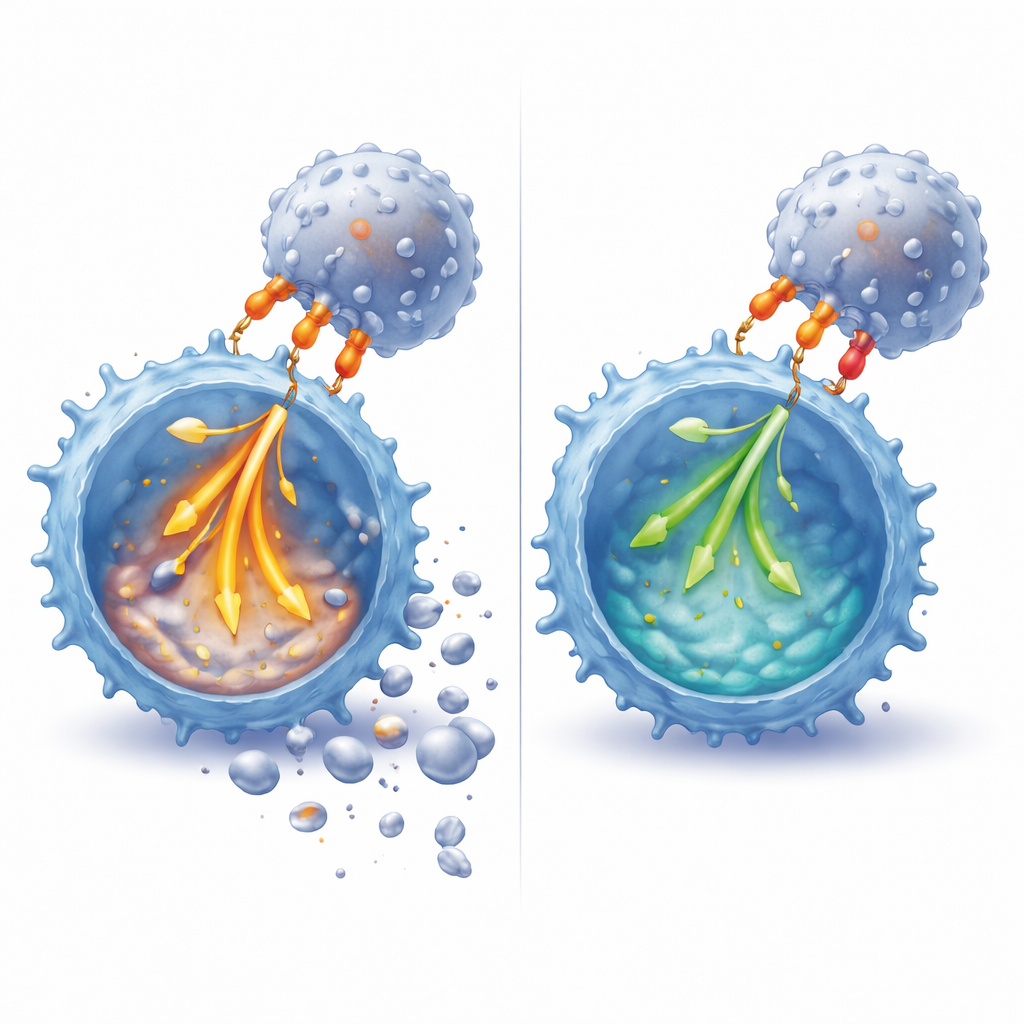
Dialing down signals and tilting survival molecules
Looking inside the cells, the researchers found that PD‑1 engagement dampened a broad range of early signaling events that follow T cell restimulation. Phosphate tags on key signaling proteins—including components directly linked to the receptor and a central relay enzyme called ERK—were noticeably reduced when PD‑L1 was present. This blunting was especially prominent when CD28 was also engaged, mirroring the larger impact on RICD in that setting. PD‑1 signaling also nudged the cell cycle, keeping more cells in the G1 “checkpoint” phase instead of pushing them aggressively into DNA replication, a state known to heighten sensitivity to death signals. At the protein level, PD‑1 shifted the balance between pro-death and pro-survival molecules: it dampened the induction of FAS ligand, a key trigger of T-cell killing, and helped preserve survivin, a factor that supports both survival and controlled division of T cells.
What these findings mean for therapies and immune health
Together, the results reveal that PD‑1 is not simply a switch that turns T cells off, but a nuanced tuner that protects newly activated human T cells from dying too soon during their expansion. By softening the strength of repeat signals through both the T cell receptor and CD28, and by favoring survival-promoting molecules over death triggers, PD‑1 allows a robust yet contained T cell army to form before the self-destruct program fully engages. For patients, this means that drugs that block PD‑1—powerful tools in oncology—may also make some T cells more vulnerable to RICD, potentially altering normal immune balance or contributing to side effects. Future therapies and cell-based treatments like CAR‑T cells may benefit from deliberately modulating PD‑1 and related pathways to preserve enough long-lived, effective T cells while still preventing harmful immune overactivity.
Citation: Lee, K.P., Elster, S., Epstein, B. et al. PD-1 protects expanding human T cells from premature restimulation-induced cell death by modulating TCR and CD28 signaling. Cell Death Dis 17, 272 (2026). https://doi.org/10.1038/s41419-026-08530-6
Keywords: PD-1 signaling, T cell survival, immune checkpoints, activation-induced cell death, cancer immunotherapy