Clear Sky Science · en
RRM1 inhibition sensitizes lung adenocarcinoma to decitabine treatment
Turning a Lukewarm Drug into a Stronger Ally
Lung cancer remains one of the deadliest cancers, and many patients eventually run out of effective treatment options. Doctors have long hoped that drugs which gently reprogram cancer cell DNA, rather than simply poisoning dividing cells, could help. One such drug, decitabine, works well in blood cancers but has disappointed in solid tumors like lung cancer. This study asks a simple, practical question with big implications: is there a way to make lung tumors finally respond to decitabine, using tools we already understand?
Why a Proven Drug Fails in Solid Tumors
Decitabine is a look‑alike version of one of the DNA building blocks. When it slips into a cell’s DNA during copying, it can erase abnormal chemical tags that silence protective genes, including tumor suppressors and immune genes. In leukemias, this helps reset cells toward a healthier state. In lung tumors, however, the drug barely works. The authors suspected that the problem was not what decitabine does, but how little of it actually makes it into the DNA of solid‑tumor cells. By measuring tiny amounts of drug built into DNA across many cancer cell lines, they confirmed that cells which incorporated more decitabine were much easier to kill with the drug.
A Cellular Gatekeeper that Blocks the Drug
To find out what limits drug entry into DNA, the researchers examined genes involved in handling nucleosides—the raw materials for DNA. One enzyme, called RRM1, stood out. RRM1 is part of a machine that converts ordinary building blocks into the active forms used to make DNA. In lung adenocarcinoma, this enzyme was unusually abundant in tumors compared with normal lung tissue, and patients with lower RRM1 levels tended to live longer. Across a panel of cancer cell lines, higher RRM1 levels went hand in hand with lower decitabine incorporation, strongly suggesting that this enzyme acts as a gatekeeper that crowds out the drug.
Disarming the Gatekeeper to Help the Drug Work
The team then asked what happens if they partially disable RRM1. Using genetic tools, they dialed down RRM1 in lung cancer cells without killing the cells outright. On its own, this reduction had only a mild effect on growth. But when combined with low doses of decitabine, the impact was dramatic: colonies of lung cancer cells shrank sharply in culture dishes, and tumors grew much more slowly in mice. Importantly, these effective doses were well tolerated, with no obvious damage to blood, liver, or kidney function in the animals. At the molecular level, blocking RRM1 allowed more decitabine to be built into DNA, leading to a stronger loss of the methyl‑adding enzyme DNMT1 and a larger drop in global DNA methylation. This, in turn, reawakened tumor‑suppressor genes that had been switched off.
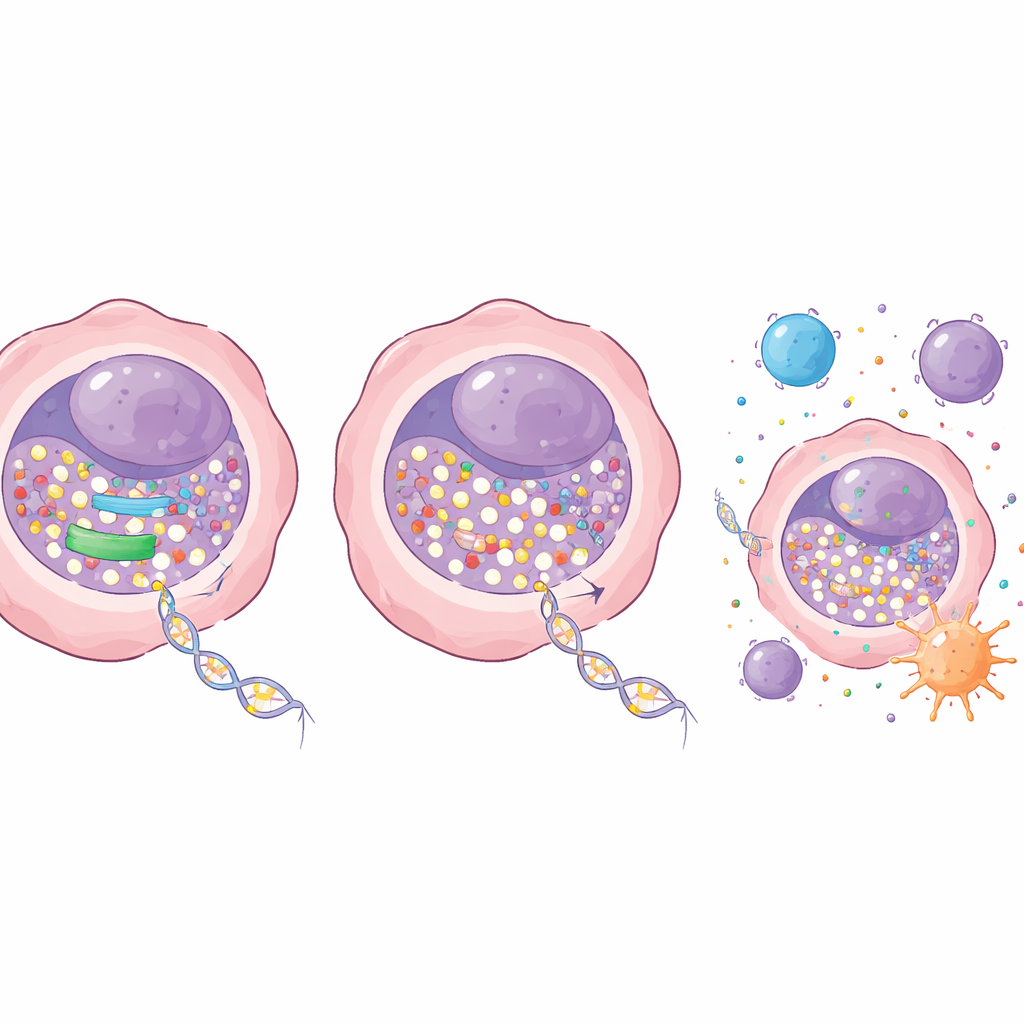
Switching On the Immune Alarm Inside Tumors
Beyond slowing cell division, the combination therapy changed how cancer cells interact with the immune system. Extra decitabine in DNA increased DNA damage signals inside the cells and pushed them toward programmed cell death. At the same time, it boosted the activity of an internal alarm system centered on the STING pathway, which senses misplaced DNA and triggers antiviral‑like immune responses. When RRM1 was blocked, decitabine more strongly activated this pathway and its downstream genes, including those that call in and stimulate immune cells. In mouse lung cancer models with intact immune systems, combining decitabine with a drug that inhibits the RRM1 enzyme produced stronger tumor control than either treatment alone, without adding obvious toxicity. The authors also found that this enzyme‑blocking strategy enhances decitabine specifically, and can actually work against a related drug, azacitidine, underscoring the need to match the right partners.
What This Could Mean for Patients
Taken together, the work paints a clear picture: an overactive DNA‑building enzyme in lung tumors limits how much decitabine reaches its target. By partially blocking this enzyme, cancer cells are forced to use more of the drug in place of their usual building blocks. That shift allows low doses of decitabine to more effectively unsilence protective genes, damage cancer cell DNA, and awaken immune defenses, all while remaining tolerable in animal models. For patients, this suggests a realistic path forward: repurposing or refining inhibitors of the RRM1 enzyme, in combination with low‑dose decitabine and potentially with modern immunotherapies, to turn a once underwhelming drug into a useful component of lung cancer treatment.
Citation: Jiang, N., Liu, J., Vaghasia, A. et al. RRM1 inhibition sensitizes lung adenocarcinoma to decitabine treatment. Cell Death Dis 17, 275 (2026). https://doi.org/10.1038/s41419-026-08522-6
Keywords: lung adenocarcinoma, decitabine, DNA methylation, ribonucleotide reductase, cancer immunotherapy