Clear Sky Science · en
PGC-1α protects against MASH via Tim23-dependent inhibition of DRP1-mediated ferroptosis
Why this matters for everyday health
Many people living with obesity or type 2 diabetes quietly develop a serious liver problem called metabolic dysfunction–associated steatohepatitis (MASH). In this condition, fat-laden liver cells become inflamed and begin to die, paving the way to scarring, cirrhosis, and liver cancer. This study uncovers a hidden self-protection system inside liver cells—centered on tiny powerhouses called mitochondria—that can either shield the liver from damage or, when it fails, accelerate disease. Understanding this internal safety switch could open doors to new treatments for one of the most common liver threats worldwide.
A closer look at a silent liver disease
MASH develops when a simple fatty liver tips into a more dangerous state marked by swollen, injured liver cells, inflammation, and eventually scar tissue. The authors examined liver samples from patients with MASH and from mouse models fed high-fat, sugar-rich or nutrient-deficient diets that mimic the human condition. They focused on a particular type of cell death called ferroptosis, in which iron and damaged fats combine to generate toxic molecules that punch holes in cell membranes. In both people and mice with MASH, liver cells showed hallmarks of this iron- and fat-driven death: excess iron deposits, distorted mitochondria, and high levels of proteins that promote lipid damage, along with low levels of proteins that normally detoxify harmful byproducts.
Evidence that blocking iron-driven cell death helps
To test whether ferroptosis is just a bystander or a driver of disease, the researchers treated mice on a high-fat diet with ferrostatin-1, a compound that specifically blocks ferroptosis. Mice receiving the blocker had less fat buildup, less iron overload, and fewer signs of inflammation and scarring in their livers. Blood tests showed improved liver function and better metabolic health, including lower cholesterol and better insulin sensitivity. In isolated mouse liver cells exposed to palmitic acid—a fat that mimics the overload seen in MASH—the same drug reduced fat accumulation, iron loading, oxidative damage, and inflammatory signals. Together, these results argue that ferroptosis is a key engine of damage in MASH and that interrupting this process can meaningfully soften the disease.
The liver’s built-in guardian inside mitochondria
The team then homed in on PGC-1α, a master regulator that helps mitochondria produce energy and cope with stress. In human MASH livers, as well as in diseased mice and stressed liver cells, PGC-1α levels were markedly lower, while a mitochondrial fission protein called DRP1 and a lipid-activating enzyme called ACSL4 were higher. Using genetically engineered mice lacking PGC-1α only in liver cells, the authors found that loss of this guardian made high-fat diets much more damaging: livers were fattier, more inflamed, more iron-loaded, and showed stronger ferroptosis signals. At the cellular level, PGC-1α deficiency boosted DRP1 activity, increased ACSL4 and iron-import proteins, and weakened the antioxidant defenses that normally keep ferroptosis in check.
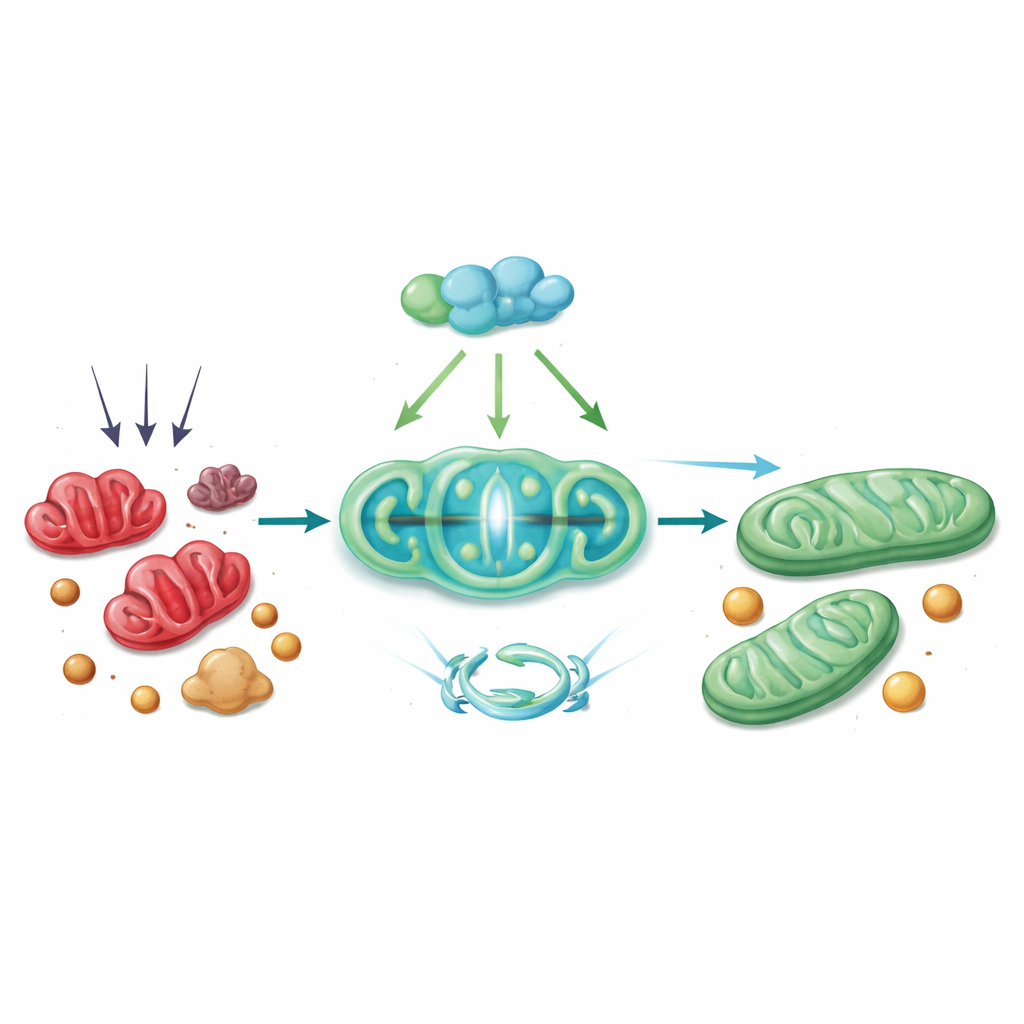
A protective chain reaction inside the cell
Mechanistically, PGC-1α acts through a chain of partners. It works with a transcription factor called Nrf1 to turn up the production of Tim23, a channel in the inner mitochondrial membrane that is essential for importing proteins and maintaining healthy structure. When Tim23 levels drop, the mitochondrial membrane potential falters, which triggers DRP1 to fragment the organelle. The study shows that with reduced Tim23, DRP1 is more active and more likely to partner with ACSL4 at the mitochondrial surface, drawing this lipid-modifying enzyme into mitochondria. There, ACSL4 helps seed the very lipid changes that make cells vulnerable to ferroptosis. Restoring PGC-1α—either in mice using a viral gene-delivery vector or in cultured hepatocytes with a CRISPR-based activator—reversed many of these steps: Tim23 went up, DRP1 and ACSL4 activity fell, mitochondria appeared healthier, and markers of ferroptosis and liver injury declined.
How this discovery could guide future therapies
To a non-specialist, the main takeaway is that the liver possesses an internal brake against iron- and fat-driven cell death, and that this brake is built into mitochondria. The PGC-1α–Tim23–DRP1–ACSL4 chain functions like a safety circuit: when PGC-1α is robust, Tim23 keeps mitochondria stable, DRP1 and ACSL4 are restrained, and liver cells are less likely to self-destruct. When this circuit fails, ferroptosis accelerates and MASH worsens. By identifying this pathway in human tissue and animal models, the study highlights two complementary strategies for future treatment—directly blocking ferroptosis, and boosting PGC-1α or Tim23 activity to steady mitochondria—offering hope for earlier and more effective interventions before irreversible liver scarring sets in.
Citation: Zhao, Y., Zhang, L., Li, B. et al. PGC-1α protects against MASH via Tim23-dependent inhibition of DRP1-mediated ferroptosis. Cell Death Dis 17, 246 (2026). https://doi.org/10.1038/s41419-026-08493-8
Keywords: fatty liver disease, mitochondria, cell death, iron metabolism, liver inflammation