Clear Sky Science · en
Targeting mitochondrial phosphatase PGAM5 alleviates ferroptosis and acute pancreatitis by upregulating NRF2-mediated FSP1 expression
Why stressed cells and sore pancreases matter
When cells are pushed too hard, they can die in ways that damage the whole body. One such form of cell death, called ferroptosis, is driven by iron and runaway chemical reactions that rust the fats in cell membranes. This process has been linked to organ injuries, including a painful and sometimes deadly condition known as acute pancreatitis. The study behind this article uncovers a key control switch inside mitochondria—the cell’s power plants—that can dial ferroptosis up or down, and shows how blocking this switch can protect the pancreas in a mouse model.
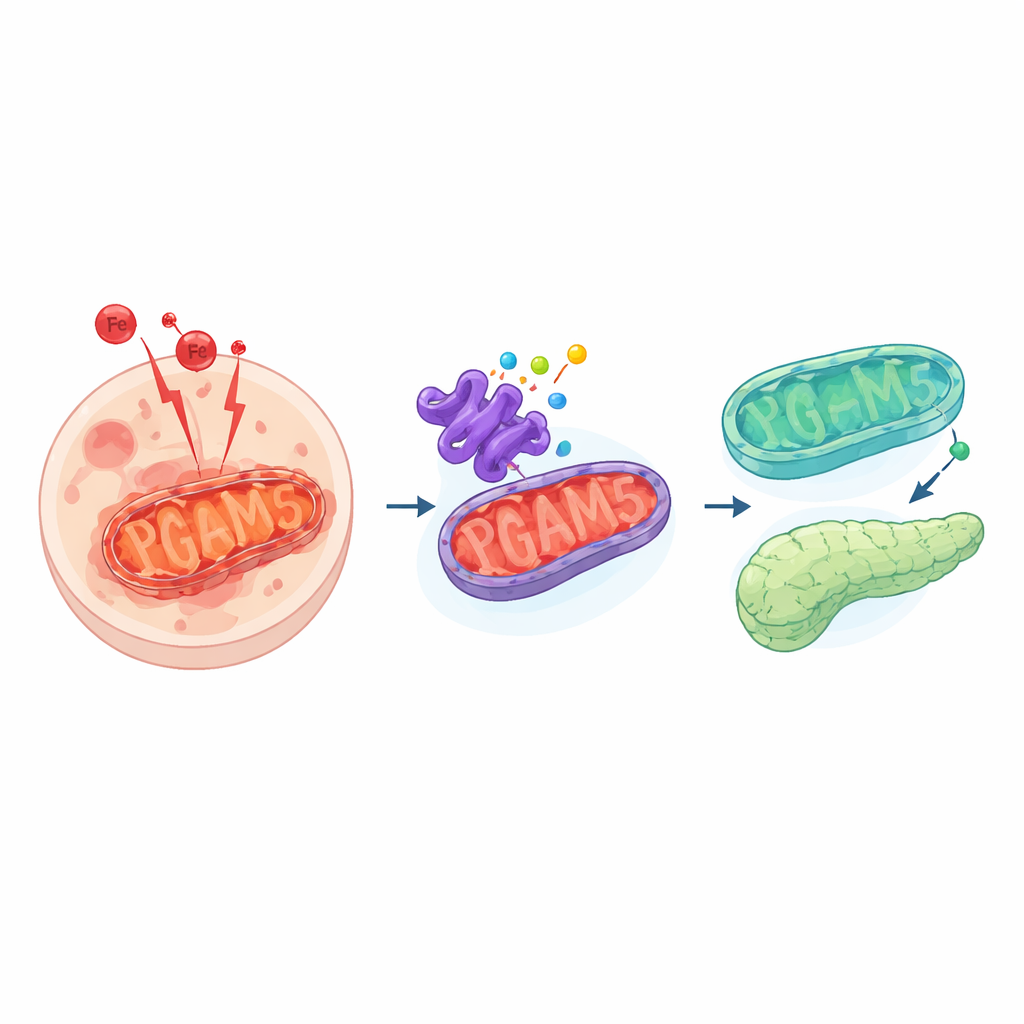
A dangerous form of cell death
Ferroptosis is different from more familiar forms of cell death like apoptosis. Instead of tidy self-destruction, cells undergoing ferroptosis experience a storm of iron-driven reactions that turn delicate fats in their membranes rancid. This creates toxic by-products and holes in membranes that ultimately kill the cell. Usually, cells keep this in check with protective systems that detoxify these reactive molecules. When those systems fail or are overwhelmed, ferroptosis can spread damage through tissues, contributing to diseases ranging from cancer to organ failure.
A mitochondrial switch in the crosshairs
The researchers focused on a protein called PGAM5, which sits on the inner face of mitochondria and acts as a signaling hub. PGAM5 helps control mitochondrial shape, responds to stress, and influences how cells handle oxidation. Surprisingly, when the team either reduced PGAM5 or forced cells to make more of it, the cells became harder to kill by ferroptosis. Chemical inhibition of PGAM5, genetic knockdown, and overexpression all lessened the build-up of damaging lipid by-products and reduced cell death triggered by a ferroptosis-inducing drug. This revealed that the system is finely tuned: both too little and too much PGAM5 push cells toward a more protected state.
Turning on an internal shield
Diving deeper, the authors discovered that PGAM5’s influence runs through a protective axis involving two other players: NRF2 and FSP1. NRF2 is a master regulator that, when active in the nucleus, switches on a broad set of antioxidant defenses. FSP1 is one of its downstream defenders that helps regenerate a fat-soluble antioxidant, blocking lipid damage at the cell membrane. When PGAM5 levels were altered, cells boosted both the message and protein levels of NRF2, and NRF2 moved more readily from the cytoplasm into the nucleus. There, it ramped up production of FSP1. Blocking FSP1 or NRF2 erased the protection, restoring sensitivity to ferroptosis, which shows that this PGAM5–NRF2–FSP1 chain is essential for the observed resilience.
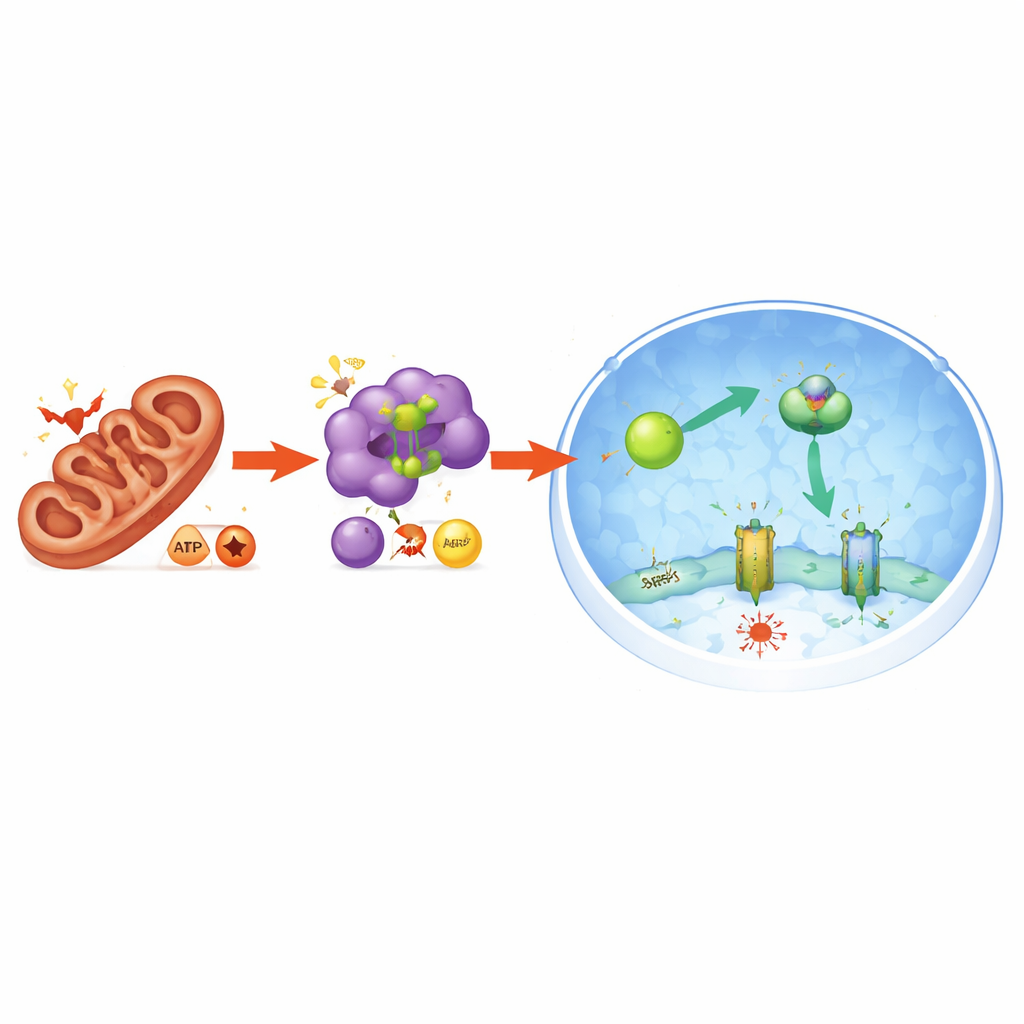
Energy stress as the hidden messenger
The study also uncovered how mitochondrial stress is translated into this protective response. Disrupting PGAM5 disturbed mitochondrial balance and shifted the cell’s energy currency: the ratio of lower-energy molecules (AMP and ADP) to high-energy ATP rose, signaling energy stress. This, in turn, activated the energy-sensing enzyme AMPK. Activated AMPK directly modified NRF2 in a way that promoted its entry into the nucleus, further boosting FSP1 production. When AMPK was removed from the system, NRF2 no longer accumulated in the nucleus, FSP1 levels fell, and cells again succumbed to ferroptosis. Thus, PGAM5 connects mitochondrial condition to a wider energy and antioxidant response that shields cells from iron-driven death.
Protecting the pancreas in living animals
To test whether this mechanism matters in a whole organ, the scientists turned to a mouse model of acute pancreatitis triggered by high doses of the amino acid arginine. In this model, the pancreas shows extensive damage, elevated levels of blood enzymes that signal tissue injury, and a surge of inflammatory molecules. Markers of lipid peroxidation—a signature of ferroptosis—also rose sharply in the pancreas. Treating mice with a PGAM5-inhibiting compound eased these symptoms: blood damage markers dropped, pancreatic tissue looked healthier under the microscope, and inflammatory signals decreased. At the same time, ferroptosis markers fell, while AMPK activity, NRF2, and FSP1 levels increased in the pancreas, matching the protective pathway seen in cell culture.
What this means for future treatments
Taken together, the work identifies PGAM5 as a central control point that links mitochondrial stress, cellular energy status, and a powerful antioxidant program that blocks ferroptosis. By dialing down PGAM5 activity, cells activate AMPK and NRF2, boost FSP1, and better withstand iron-driven lipid damage. In mice, this strategy lessens pancreatic injury in acute pancreatitis. For a lay reader, the message is that researchers have found a new internal “circuit breaker” that can prevent a destructive form of cell death. While much remains to be done before clinical use, targeting PGAM5 or its downstream partners could open new avenues to treat conditions where ferroptosis and mitochondrial failure play a harmful role.
Citation: Ma, S., Qin, J., Luan, J. et al. Targeting mitochondrial phosphatase PGAM5 alleviates ferroptosis and acute pancreatitis by upregulating NRF2-mediated FSP1 expression. Cell Death Dis 17, 252 (2026). https://doi.org/10.1038/s41419-026-08484-9
Keywords: ferroptosis, mitochondria, acute pancreatitis, oxidative stress, cell death