Clear Sky Science · en
Semaphorin 6D drives anti-tumor type I interferon responses to reprogram the tumor microenvironment in colorectal cancer
Why this research matters for people with colon cancer
Colorectal cancer is one of the leading causes of cancer deaths worldwide, in part because many tumors resist today’s treatments, including cutting-edge immunotherapy drugs. This study uncovers a natural "braking system" inside colon tumors that is often switched off, and shows how turning it back on can invite the immune system back into the fight. Understanding this hidden control switch could help doctors better predict outcomes and design combination therapies that make immunotherapy effective for many more patients.
A silent guardian inside tumor cells
At the center of the work is a molecule called Semaphorin 6D (SEMA6D), originally known for guiding nerve growth and shaping the developing heart. The researchers found that SEMA6D actually behaves like a tumor suppressor in colorectal cancer: in healthy colon tissue it is present, but in cancerous tissue its levels are markedly reduced. Across multiple patient datasets and tumor samples, low SEMA6D was linked to larger tumors, deeper invasion, more metastases, and significantly poorer survival. This pattern held even when other clinical factors were accounted for, indicating that SEMA6D is an independent marker of how aggressive a colorectal tumor is.
How tumors switch off this protection
The team then asked why SEMA6D is so often missing in tumors. They discovered that the gene is frequently shut down by a chemical modification called promoter hypermethylation—extra chemical tags added to the gene’s control region that act like molecular duct tape over a light switch. Using detailed DNA mapping, they showed that key stretches of the SEMA6D control region are heavily methylated in cancer cells but not in normal colon cells. When they treated cancer cells with a demethylating drug used in blood cancers, the methyl marks were stripped away and SEMA6D production was restored. The lowest SEMA6D levels were found in colon cancer subtypes already known for heavy DNA methylation, high genetic instability, and strong tendencies toward spread, reinforcing the link between this silencing mechanism and aggressive disease.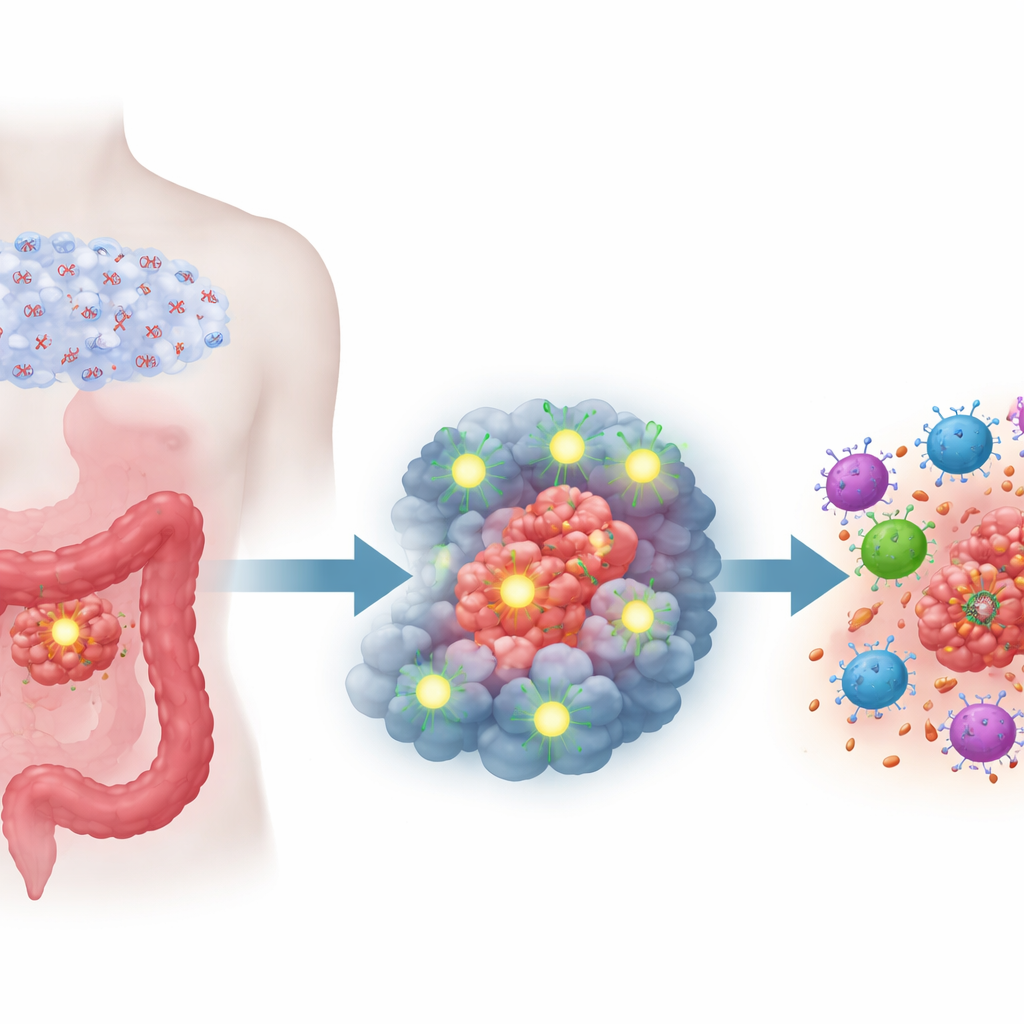
From growth blocker to immune booster
Restoring SEMA6D changed tumor behavior on two levels. First, at the cancer cell level, forcing cells to make more SEMA6D slowed their growth, reduced their ability to migrate and invade, and reversed features of epithelial–mesenchymal transition, a program that helps tumors spread. In dishes and three-dimensional organoids grown from patient tumors, cells with more SEMA6D formed fewer, smaller colonies and showed more signs of programmed cell death. In mice, tumors engineered to overproduce SEMA6D grew more slowly and produced fewer lung and liver metastases, whereas knocking SEMA6D down had the opposite effect. Second, at the immune level, SEMA6D-rich tumors in immunocompetent mice contained many more CD4 and CD8 T cells—the main attack forces of adaptive immunity—while SEMA6D-poor tumors were relatively empty of these defenders. When the researchers depleted T cells, the growth-slowing effect of SEMA6D largely disappeared, showing that much of its power comes from rallying the immune system.
Decoding the internal alarm pathway
Diving deeper, the study mapped the molecular steps that connect SEMA6D to immune activation. On the surface of tumor cells, SEMA6D signals through a partner receptor called Plexin A4. Inside the cell, this duo physically interacts with a protein named IRF9, a key component of the machinery that responds to type I interferons—the same antiviral alarm signals that cells use to fight infections. When SEMA6D is present and Plexin A4 is intact, IRF9 and its partners become activated, switch on suites of interferon-stimulated genes, and help the tumor cell broadcast signals that attract and arm T cells. Removing SEMA6D or Plexin A4 breaks this chain and mutes the alarm; restoring IRF9 can partially rescue the effect. In mice, tumors with active SEMA6D–Plexin A4–IRF9 signaling had more infiltrating T cells and lower levels of the proliferation marker Ki-67, consistent with stronger immune pressure on the cancer.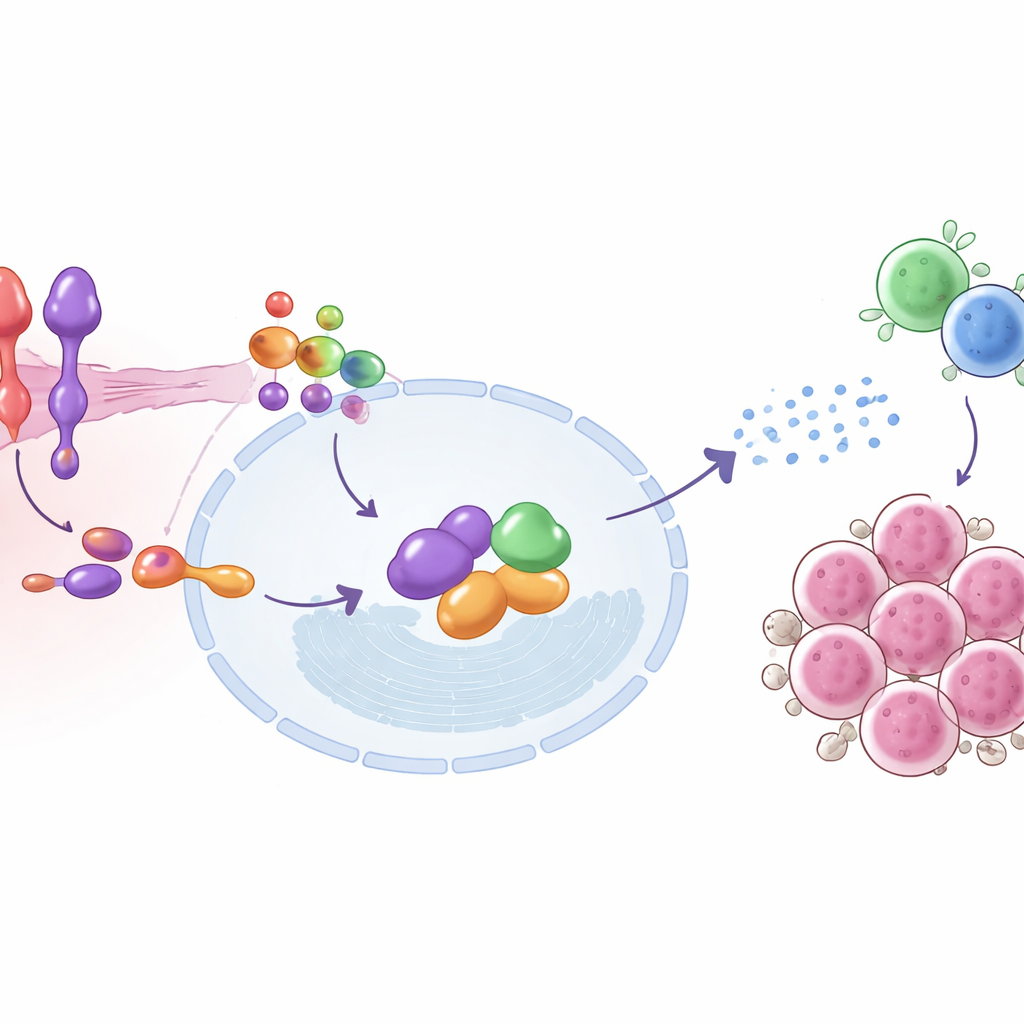
Reawakening immunity with combination therapy
Because SEMA6D is silenced by methylation, the authors tested whether a hypomethylating drug could reactivate it in living tumors and thereby improve responses to immune checkpoint blockade. In mouse colon tumors treated with decitabine followed by an anti–PD-1 antibody, tumors grew far more slowly than with either treatment alone. The combination increased SEMA6D levels, boosted interferon pathway activity, reduced cell proliferation, and enhanced T cell infiltration. These results suggest that, by peeling off methylation “locks” from immune-relevant genes like SEMA6D, epigenetic drugs can turn immunologically “cold” tumors into “hotter” ones that are more vulnerable to checkpoint inhibitors.
What this means for future care
To a layperson, the conclusion is that some colon cancers hide from the immune system by chemically shutting down a built-in danger signal. This work identifies SEMA6D as both that signal and a promising handle for therapy. Measuring SEMA6D and its methylation status could help classify tumors, forecast outcomes, and guide treatment choices. Just as important, the study offers a clear biological rationale for combining DNA-demethylating agents with immunotherapy to reawaken immune surveillance in patients whose tumors currently do not respond. While clinical trials are still needed, this strategy could one day broaden the benefits of immunotherapy to a much larger group of people with colorectal cancer.
Citation: Shi, W., Zhang, F., Sun, WQ. et al. Semaphorin 6D drives anti-tumor type I interferon responses to reprogram the tumor microenvironment in colorectal cancer. Cell Death Dis 17, 256 (2026). https://doi.org/10.1038/s41419-026-08478-7
Keywords: colorectal cancer, tumor microenvironment, epigenetic therapy, type I interferon, tumor immunology