Clear Sky Science · en
Loss of function variants in HPDL impair human cortical development via alterations of mitochondrial function
Why tiny cell engines matter for growing brains
Most people think of genetics and wiring when they picture how the brain develops. This study shows that another, often overlooked factor—the small power plants inside our cells called mitochondria—can also shape how our brains form. By studying rare childhood movement disorders linked to a gene named HPDL, the authors reveal how faulty energy production can shrink the developing cortex, the brain region crucial for movement, thinking, and behavior.
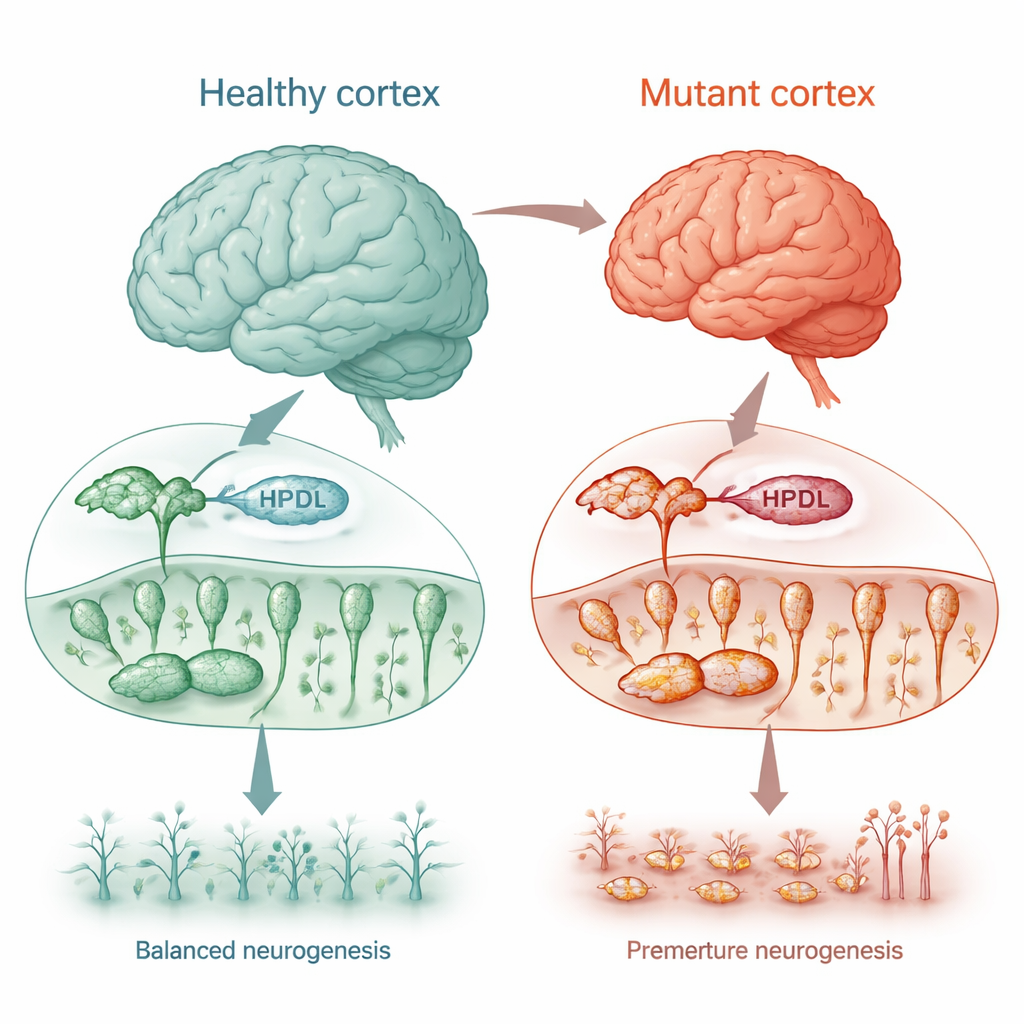
A rare movement disorder as a window into brain growth
Some children with changes in the HPDL gene develop hereditary spastic paraplegia, a condition that causes stiffness and weakness in the legs, along with seizures, developmental delay, and in severe cases a smaller-than-normal brain (microcephaly). Although the HPDL protein was known to sit in mitochondria, its exact job—and why its loss damages the brain—was unclear. The researchers used several human cell models, including nerve-like tumor cells and brain cells grown from patient skin samples, to test whether HPDL is needed for normal brain development and mitochondrial health.
What happens when HPDL is switched off
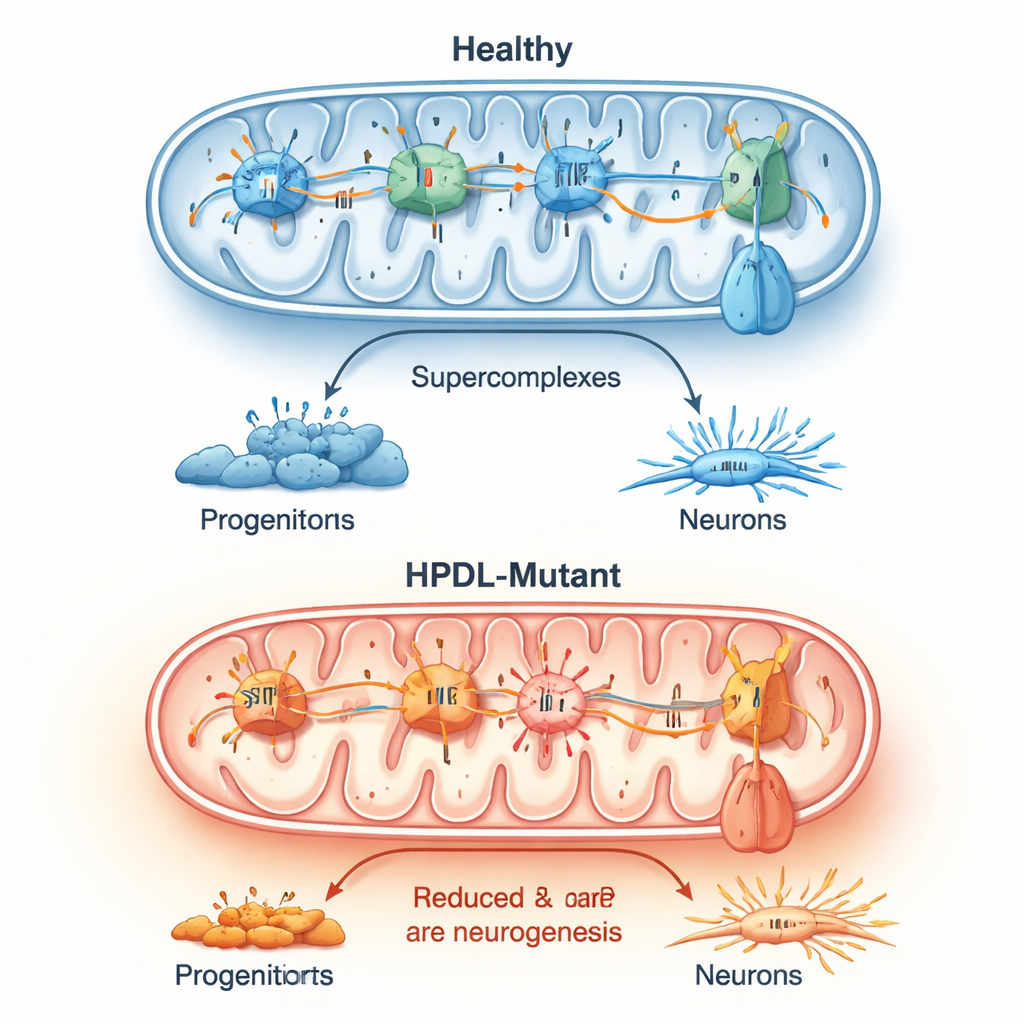
First, the team turned off HPDL in a human neuroblastoma cell line using CRISPR gene editing. Without HPDL, these cells lost the full-length protein and showed clear mitochondrial problems. The large assemblies of respiratory chain proteins that normally work together to generate energy were disrupted, and key components involved in oxygen use were reduced. The cells burned less oxygen, produced less energy-linked respiration, and generated more reactive oxygen species—harmful by-products often called “oxidative stress.” Yet the total number of mitochondria did not decrease, and levels of coenzyme Q10, a vital molecule for energy transfer, were actually higher, suggesting a qualitative—not just quantitative—defect in mitochondrial function.
Brain tissue in a dish reveals early overproduction of neurons
To see how HPDL loss affects real human brain development, the researchers reprogrammed skin cells from four affected children into induced pluripotent stem cells and then coaxed them into forming cortical brain cells and three-dimensional “mini-brains” (organoids). Early in development, at a stage when most cells should still be dividing as neural progenitors, HPDL-mutant cultures already contained more mature neurons and fewer progenitors. Gene activity profiles backed this up: pathways that drive neuron formation were switched on too soon, while those that keep cells in a dividing state were dialed down. In organoids, this premature shift from building blocks to finished neurons led to much smaller brain-like structures, echoing the microcephaly seen in the sickest children.
Broken power plants and stressed cells
Closer inspection showed that HPDL-mutant brain cells had impaired oxidative phosphorylation—the main way mitochondria make energy. Enzyme staining revealed weaker activity of a key mitochondrial complex, while other measures showed altered electrical charge across the mitochondrial membrane. In many mutant cells, a crucial enzyme that normally makes ATP appeared to work in reverse to prop up this membrane charge, a sign of deep metabolic distress. Across patient lines, reactive oxygen species were consistently elevated, and the normal large assemblies of respiratory chain proteins were less well formed. These mitochondrial changes closely tracked with the timing and degree of premature neuron production.
Testing ways to ease the stress
Because oxidative stress and disturbed coenzyme Q10 chemistry seemed central, the team tested whether treatments targeting these problems could slow down the rush into neuron formation. They exposed early cortical cultures to two antioxidants and to 4-hydroxybenzoate, a small molecule related to coenzyme Q10 synthesis. In several patient-derived lines, these compounds partly reduced premature neurogenesis, but the response depended on the exact HPDL mutation. Some lines responded mainly to antioxidants, others to the coenzyme Q10 precursor, and one did not respond at all. This mutation-specific pattern suggests that personalized treatment strategies may be needed for HPDL-related disorders.
What this means for children and future therapies
In simple terms, this study shows that HPDL acts as a guardian of the brain’s building blocks during early development. When HPDL fails, mitochondria become inefficient and overly stressed, pushing progenitor cells to become neurons too soon. The pool of dividing cells is drained, the cortex cannot reach its full size, and wiring patterns are altered, contributing to movement problems and other symptoms. The partial rescue seen with antioxidants and coenzyme Q10–related compounds hints that adjusting cellular energy balance and oxidative stress could someday help children with HPDL mutations, and perhaps others with mitochondrial forms of brain disease.
Citation: Baggiani, M., Desbats, M.A., Naef, V. et al. Loss of function variants in HPDL impair human cortical development via alterations of mitochondrial function. Cell Death Dis 17, 237 (2026). https://doi.org/10.1038/s41419-026-08476-9
Keywords: HPDL, mitochondria, cortical development, microcephaly, oxidative stress