Clear Sky Science · en
c-Myc/GRPEL1 maintains fatty acid synthesis via FASN to support PDAC cell proliferation
Why Cancer Cells’ Fat Cravings Matter
Pancreatic cancer is one of the deadliest cancers, in part because its cells are experts at reprogramming their own metabolism to survive and grow. This study looks at a surprising part of that story: how pancreatic tumor cells ramp up their internal “fat factories” to fuel relentless growth, and how blocking this process might open up new treatment options for patients.
A Tough Cancer with a Metabolic Edge
Most pancreatic cancers are a type called pancreatic ductal adenocarcinoma (PDAC), which is usually detected late and responds poorly to today’s therapies. PDAC cells live in a harsh environment with little oxygen and scarce nutrients, yet they thrive by rewiring how they use sugar, fats, and other fuels. Their mitochondria—tiny power plants inside cells—play a central role in this rewiring. To keep these power plants running, cells constantly monitor and repair thousands of mitochondrial proteins, a process known as mitochondrial protein quality control. Until now, it was unclear how this quality control machinery links to the way pancreatic tumors feed themselves.
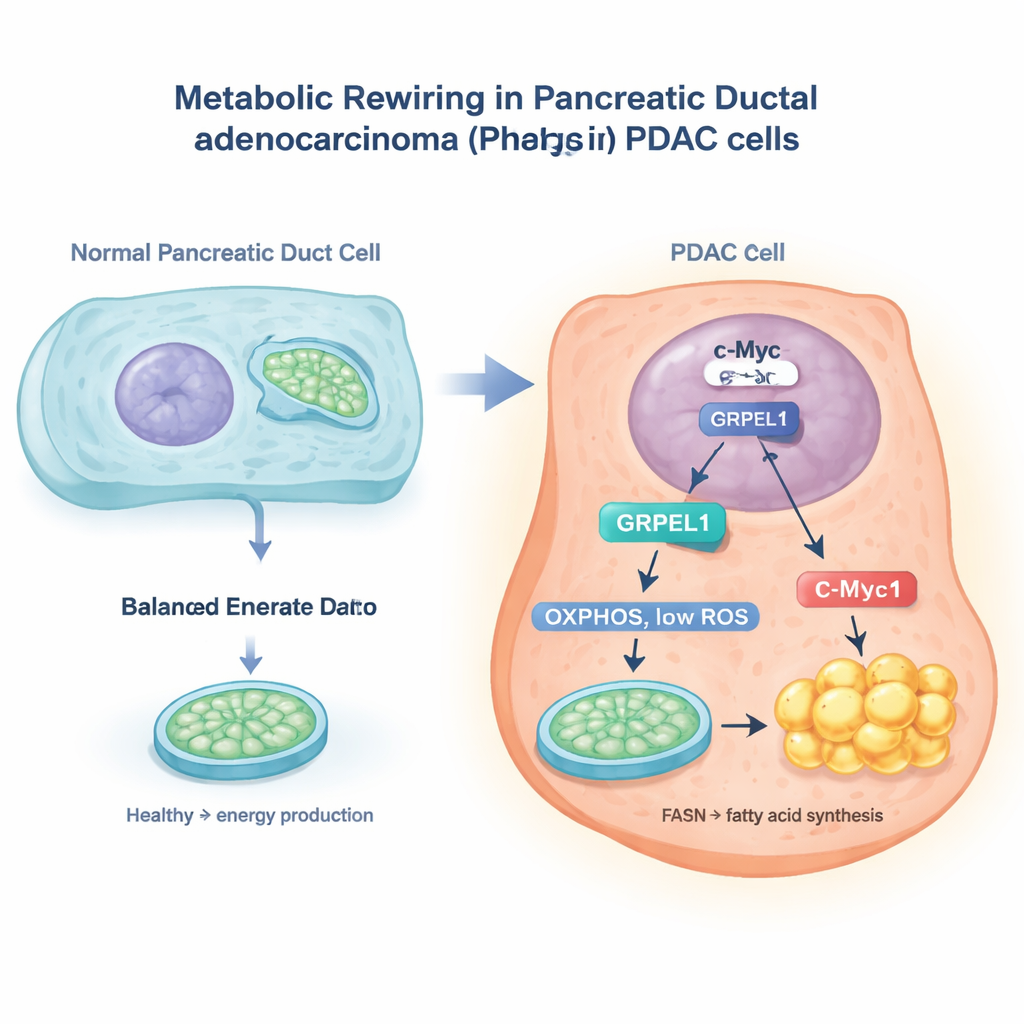
The c-Myc Switch and a Mitochondrial Helper
The researchers used large cancer databases and experiments in pancreatic cancer cells to home in on a protein called GRPEL1, a helper involved in mitochondrial protein handling. They discovered that a well-known cancer gene, c-Myc, acts like a molecular switch in the cell nucleus that turns on the GRPEL1 gene. When c-Myc levels were reduced, GRPEL1 levels fell; when c-Myc was boosted, GRPEL1 rose. Patients’ tumor samples also showed that c-Myc and GRPEL1 tend to be high together, and both were linked to worse outcomes. In lab-grown PDAC cells, lowering GRPEL1 slowed cell division and colony growth, while adding extra GRPEL1 helped cells proliferate faster, especially when c-Myc was otherwise blocked.
From Mitochondria to Fat Production
Digging deeper, the team found that GRPEL1 does more than just keep mitochondrial proteins in line. When GRPEL1 was depleted, mitochondria in PDAC cells became less efficient at producing energy, lost their normal shape, and released more reactive oxygen species (ROS)—chemically reactive byproducts sometimes called cellular “rust.” This surge in ROS had a knock-on effect: it lowered the levels of fatty acid synthase (FASN), a key enzyme that builds new fatty acids inside cells. With FASN dialed down, the cells made fewer fats, stored fewer lipids, and their growth slowed. When the researchers mopped up ROS with an antioxidant, FASN levels bounced back, showing that the GRPEL1–FASN link is driven by ROS. Interestingly, c-Myc itself did not appear to directly switch on the FASN gene in this system, but instead influenced FASN indirectly through GRPEL1 and mitochondrial stress.
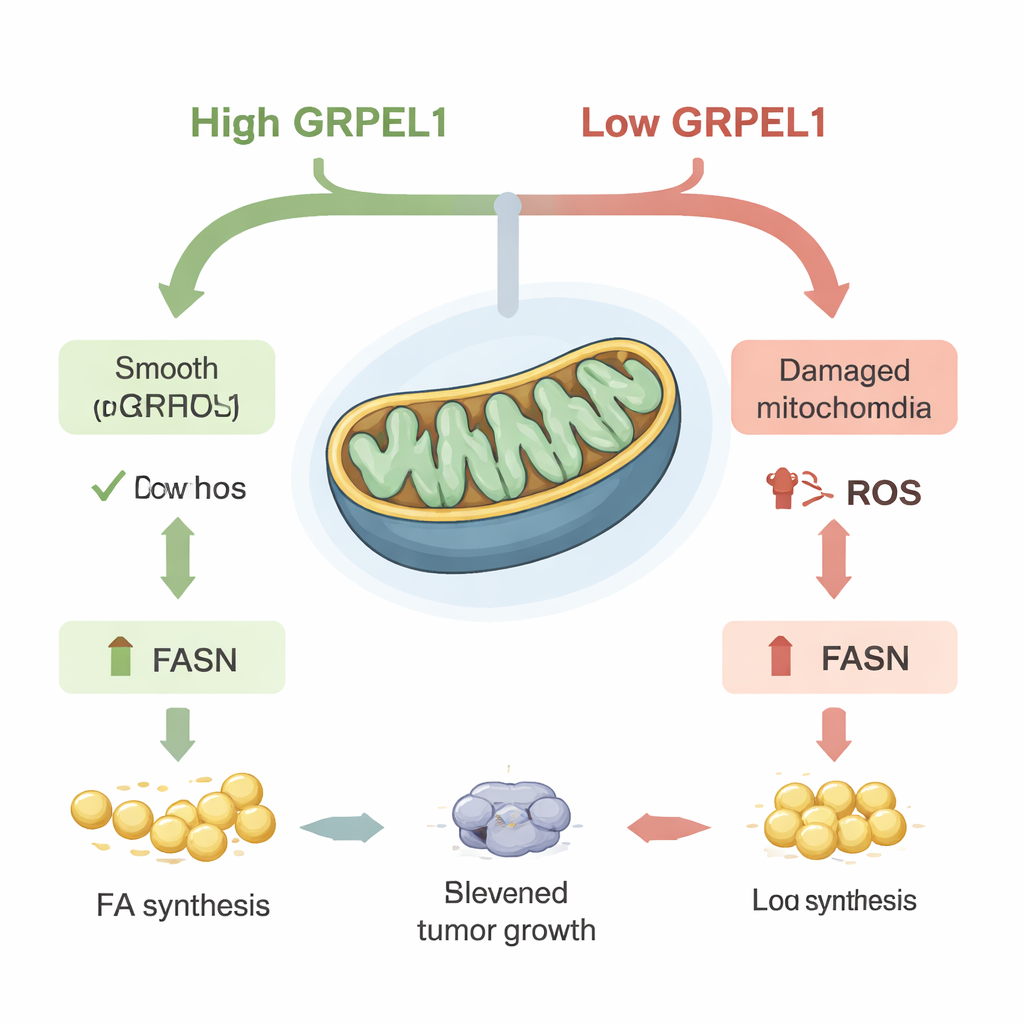
Fat as Fuel for Tumor Growth
Because FASN sits at the heart of fat-building, the scientists asked whether restoring it could rescue tumor cell growth when GRPEL1 was low. In cell cultures, forcing cells to make more FASN partly reversed the slowdown in growth and colony formation caused by loss of GRPEL1. The same was true in mice: tumors formed from GRPEL1-depleted cells grew more slowly, but reintroducing FASN revived both tumor size and fat content. Detailed analyses of metabolites and lipids showed broad drops in many fat-related molecules when GRPEL1 or c-Myc were reduced. Importantly, supplying extra fatty acids or a lipid mixture from the outside partially restored growth in both cancer cell lines and patient-derived pancreatic cancer organoids—mini-tumors grown in 3D culture—suggesting that the main problem was loss of newly made fats.
Turning a Vulnerability into a Therapy
Altogether, the work paints a clear picture: in pancreatic cancer, c-Myc turns up GRPEL1, which helps mitochondria run smoothly and keeps ROS in check. This calm mitochondrial environment allows cells to maintain high levels of FASN, pumping out new fatty acids that serve as building blocks for membranes, energy storage, and growth signals. When GRPEL1 is blocked, mitochondria falter, ROS rises, FASN falls, and the cancer cells struggle to grow—an effect that can be partly bypassed if fats are supplied from outside. For lay readers, the takeaway is that pancreatic tumors rely on an internal “fat factory” circuit, driven by c-Myc, GRPEL1, and FASN. Drugs that disrupt this fatty acid–making axis, especially in tumors where it is highly active, could offer a promising new way to starve pancreatic cancer cells while leaving normal tissues less affected.
Citation: Wang, J., Zhang, L., Chen, K. et al. c-Myc/GRPEL1 maintains fatty acid synthesis via FASN to support PDAC cell proliferation. Cell Death Dis 17, 205 (2026). https://doi.org/10.1038/s41419-026-08439-0
Keywords: pancreatic cancer, tumor metabolism, fatty acid synthesis, mitochondria, c-Myc