Clear Sky Science · en
Mutant TDP-43 drives impairments in axonal transport and glycolysis in a mouse stem-cell-derived motor neuron model of amyotrophic lateral sclerosis (ALS)
Why this research matters for people with ALS
Amyotrophic lateral sclerosis (ALS) is a fatal disease that gradually paralyzes people by killing the nerve cells that control muscles. Most patients show damage involving a protein called TDP-43, but scientists still do not fully understand how this damage actually harms motor neurons. This study uses a carefully engineered mouse stem-cell model to pinpoint some of the earliest problems caused by a disease-linked version of TDP-43, offering clues that could help shape future treatments.
Building motor neurons in a dish
To explore ALS in a controlled setting, the researchers started with mouse embryonic stem cells and coaxed them to become motor neurons, the long, cable-like cells that send signals from the spinal cord to muscles. They inserted a single extra copy of the human TDP-43 gene into these cells, either in its normal form or carrying a specific ALS-associated mutation called M337V. A fluorescent tag allowed the team to track the human protein inside the cells. By day 20 in culture, both normal and mutant cells had matured into motor neurons that expressed typical markers, formed branching networks, and made synapse-like connections, closely mimicking neurons found in the nervous system. 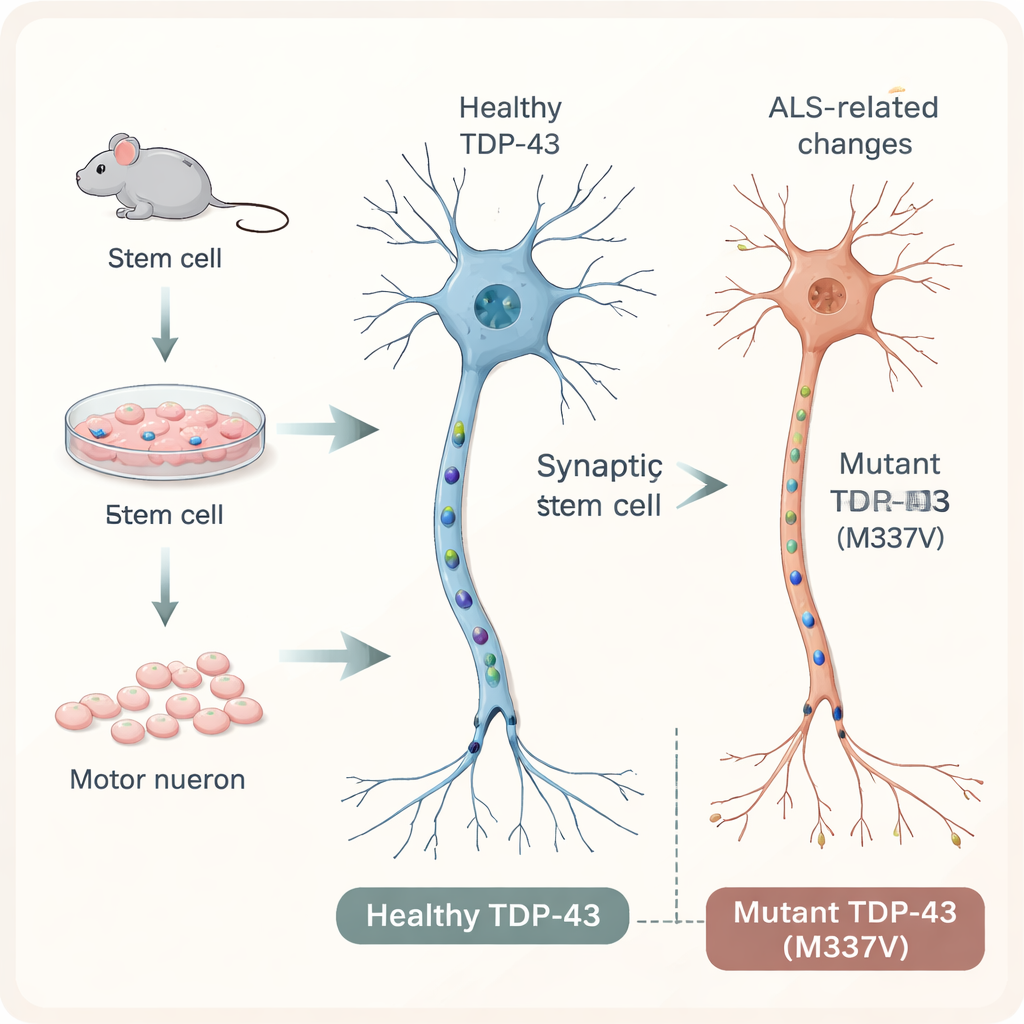
Hidden damage without visible protein clumps
In people with ALS, TDP-43 often moves from its usual position in the nucleus to the cell’s outer compartment and forms clumps, a classic pathological hallmark seen under the microscope. Surprisingly, in this stem-cell model, the mutant TDP-43 did not show increased mislocalization or aggregation compared with the normal version. Most of the protein remained where it should be. Yet the neurons were clearly less healthy: cultures with the mutant protein had fewer cell bodies, smaller networks of nerve fibers, and reduced overall viability. This suggests that serious harm to motor neurons can occur before, or even without, the dramatic protein clumps seen in patient brains and spinal cords.
Traffic jams along the nerve “highways”
Motor neurons rely on fast, efficient transport systems that move vital cargo up and down their long axons. Using microfluidic devices that isolate axons in tiny channels, the team followed the movement of signaling packets (endosomes) and energy-producing mitochondria in live cells. In neurons with mutant TDP-43, these cargoes still moved mostly in the correct directions, but they traveled more slowly. Signaling endosomes showed reduced speeds when moving back toward the cell body, and mitochondria moved more sluggishly in both directions, with more time spent paused in place. Importantly, the basic machinery that drives this motion—the motor proteins that walk along internal tracks—did not appear to change in amount, hinting that the problem lies in how this machinery functions rather than how much of it is present.
Energy shortfalls in sugar burning, but not in mitochondria
Because axonal transport is energy-hungry, the researchers tested how these neurons make and use energy. They measured two main sources: mitochondrial respiration, which burns fuel using oxygen, and glycolysis, which breaks down sugar in the surrounding fluid. Mitochondria looked normal in number, shape, and membrane potential, and their overall ability to produce energy appeared unchanged in the mutant cells. In contrast, the neurons with mutant TDP-43 showed a clear drop in baseline glycolysis. Previous work has shown that local glycolysis along axons can supply “on-board” fuel for fast transport of vesicles. The reduced sugar-burning capacity seen here may therefore contribute to the slowed movement of cargoes, adding another layer of stress to already vulnerable motor neurons.
What this means for future ALS therapies
Taken together, the study shows that even low levels of ALS-linked mutant TDP-43 are enough to make motor neurons more fragile, slow the movement of essential cargo along their axons, and dampen their ability to generate energy from sugar—all without the obvious protein clumps that pathologists usually look for. For non-specialists, the key message is that early, subtle changes in cellular “traffic flow” and energy use may set the stage for later, more dramatic damage in ALS. This highlights axonal transport and cellular energy pathways, particularly glycolysis, as promising targets for therapies aimed at protecting motor neurons before irreversible degeneration occurs. 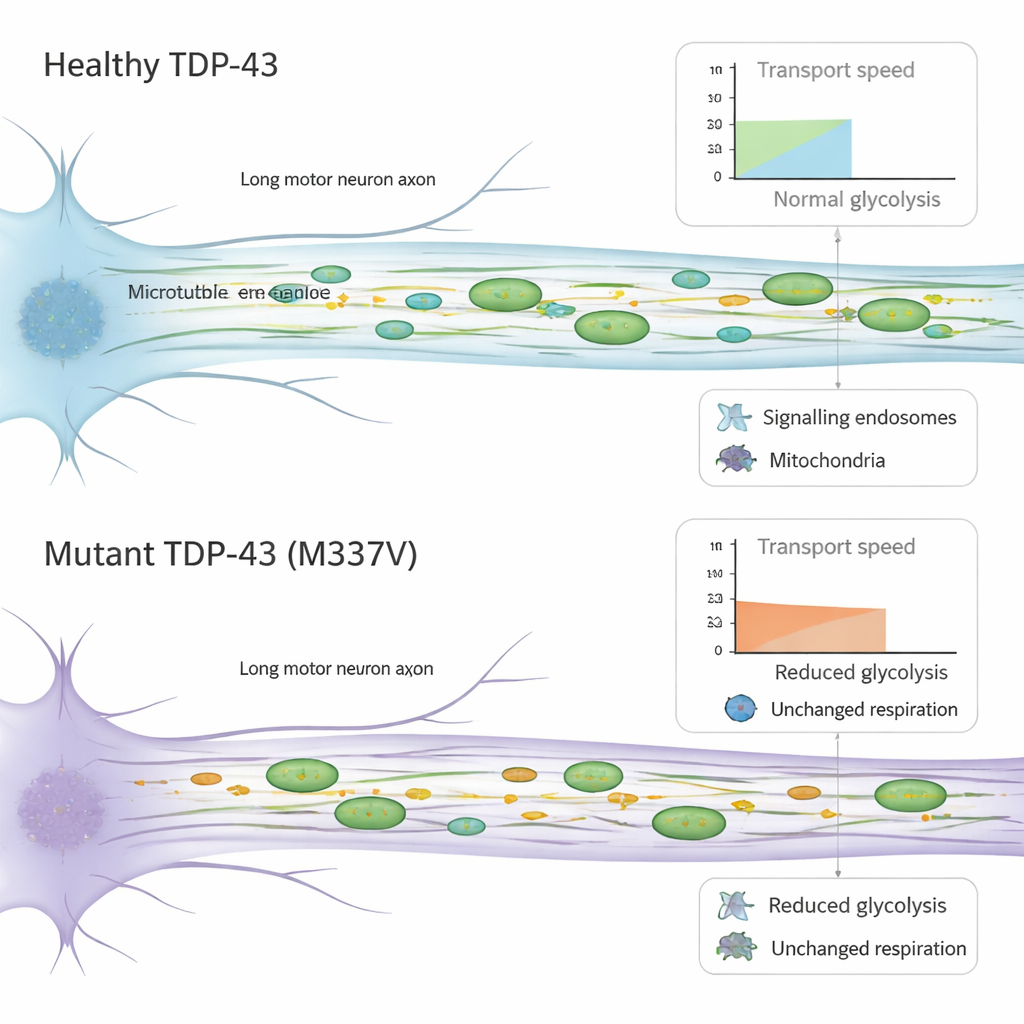
Citation: Carroll, E., Scaber, J., Pasniceanu, IS. et al. Mutant TDP-43 drives impairments in axonal transport and glycolysis in a mouse stem-cell-derived motor neuron model of amyotrophic lateral sclerosis (ALS). Cell Death Dis 17, 193 (2026). https://doi.org/10.1038/s41419-026-08437-2
Keywords: amyotrophic lateral sclerosis, TDP-43, motor neurons, axonal transport, cellular energy