Clear Sky Science · en
BCL-xL as a therapeutic target in cetuximab-refractory colorectal cancer
Why this matters for people with colon cancer
Colorectal (colon and rectal) cancer is one of the most common cancers worldwide, and many patients with advanced disease receive a targeted antibody drug called cetuximab. This drug can shrink tumors at first, but in most patients the cancer finds ways to escape within months, leaving doctors with few good options. This study asks a pressing question: when colon tumors stop responding to cetuximab, is there another weak point that new medicines can exploit to make the cancer cells die?
When a targeted drug stops working
Cetuximab works by blocking a surface antenna on cancer cells called the epidermal growth factor receptor (EGFR), which helps drive their growth. The team created a laboratory model of resistance by exposing a sensitive colorectal cancer cell line (LIM1215) to gradually increasing doses of cetuximab over six months. Two independently derived resistant cell populations emerged that kept growing even in high drug levels, yet looked just as healthy and fast-growing as the original cells when the drug was removed. Importantly, the resistant cells still carried the drug’s target on their surface and cetuximab could still bind it, suggesting the cancer had not simply “hidden” or altered the receptor.
Resistant cells reroute their growth signals
To understand how the cells were bypassing cetuximab, the researchers examined key growth pathways inside the cell. In parental cells, cetuximab normally dampened the MAPK pathway, a major engine of cell division. In the resistant cells, MAPK activity stayed high even when EGFR was blocked, showing that the growth signal had been uncoupled from the original receptor. Sequencing of cell RNA revealed new activating mutations in another RAS gene, HRAS, in subpopulations of the resistant cells, but not in other usual suspects such as KRAS, NRAS or BRAF. Attempts to shut down this rerouted signaling with a MEK inhibitor drug (which acts downstream of RAS) reduced growth only modestly. This underscored that, rather than chasing every new mutation, it might be more effective to attack cellular life-or-death machinery shared across different resistant clones. 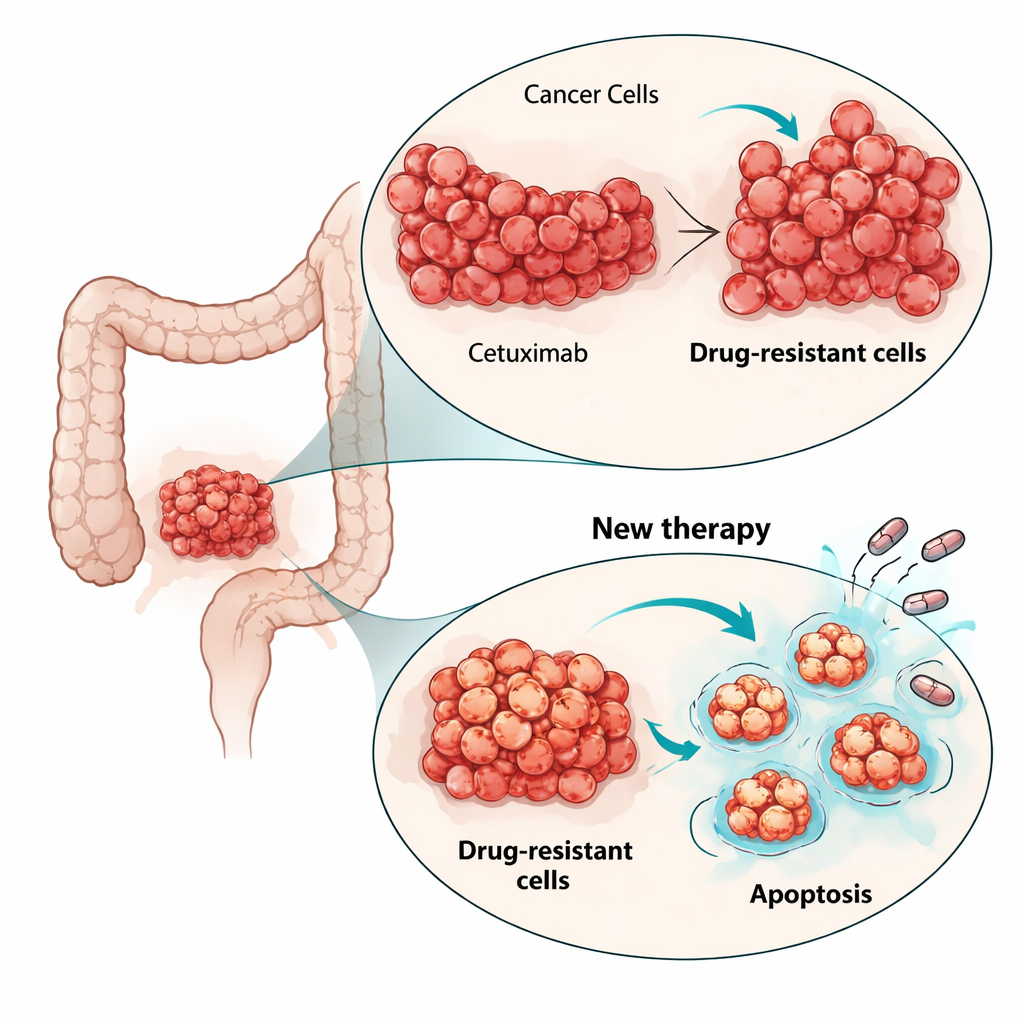
Hitting cancer at its life-support system
The scientists turned their focus to apoptosis, the built-in cell suicide program that cancers often suppress. Gene expression analysis showed that apoptosis-related pathways were altered in the resistant cells. In particular, the anti-death protein BCL-xL was higher in one resistant population and modestly increased in the other, while another survival protein, MCL-1, was also present. The team tested small-molecule drugs called BH3 mimetics that are designed to block these survival proteins and free up the death machinery. In two-dimensional cell cultures, all three cell lines—parental and resistant—were sensitive to drugs blocking BCL-xL or MCL-1, but, strikingly, lower doses killed the cetuximab-resistant cells more effectively. Adding a low dose of the proteasome inhibitor bortezomib, which helps accumulate pro-death signals, further boosted killing, especially when combined with the MCL-1 blocker.
From flat dishes to 3D mini-tumors and patient tissues
Because flat cell layers cannot fully mimic tumors in the body, the team next grew the cells as three-dimensional spheroids embedded in a gel, better reflecting the architecture and drug penetration challenges of real tumors. Again, blocking BCL-xL or MCL-1 reduced spheroid viability, and combining these drugs with bortezomib caused dramatic drops in metabolic activity and clear signs of cell death. To test whether this vulnerability exists in more realistic human tumor material, they used thin slices of cetuximab-resistant colorectal cancers grown in mice from patient tumors (patient-derived xenografts). These models were all KRAS wild-type like the original LIM1215 cells but carried diverse additional mutations, including in BRAF and TP53, mirroring the genetic variety seen in the clinic. 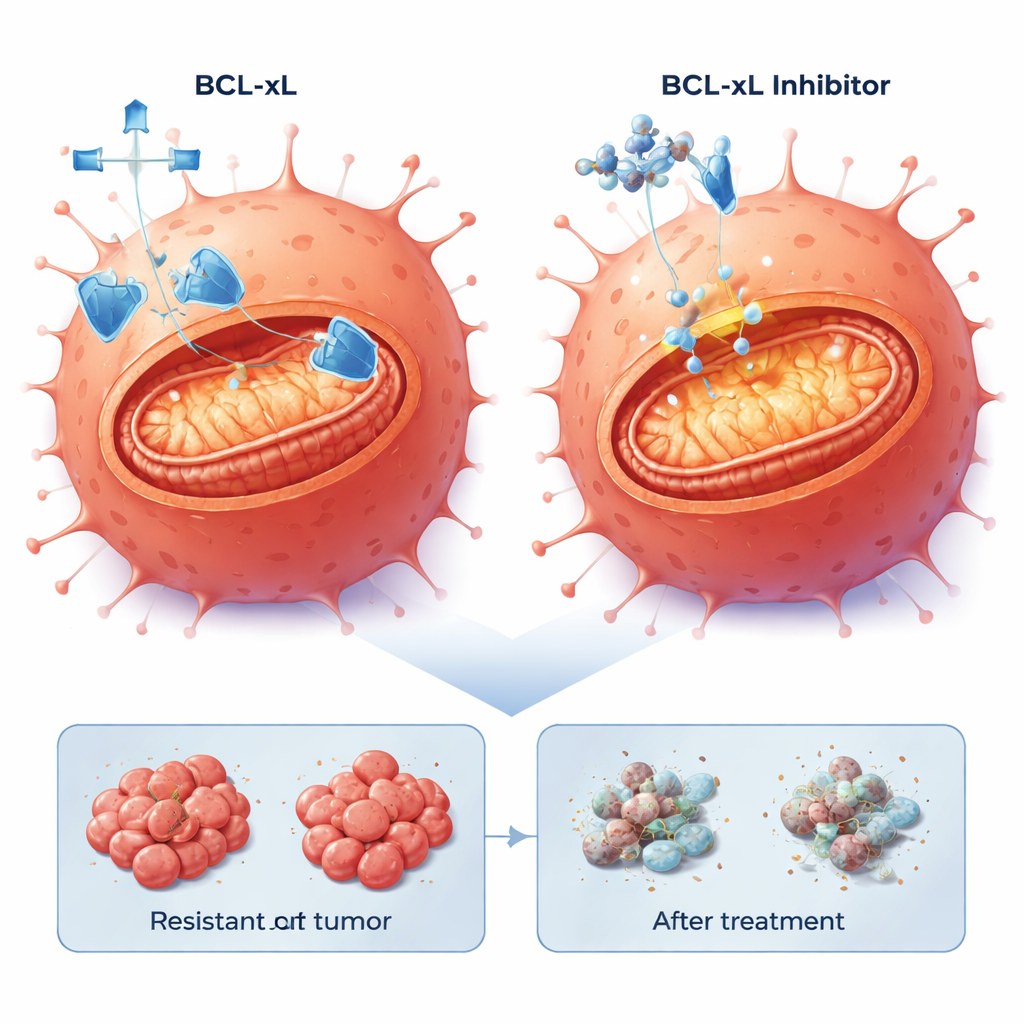
Targeting BCL-xL works across diverse resistant tumors
In these patient-derived tumor slices, the combination of a BCL-xL inhibitor with low-dose bortezomib consistently triggered robust cell death in 20–40% of tumor cells across four different models, including those with aggressive BRAF mutations. By contrast, blocking MCL-1 with bortezomib produced strong effects only in a subset of tumors. Importantly, the ability of resistant cells to undergo apoptosis was preserved: once the BCL-xL safety net was removed, the internal death program could still be activated, regardless of the specific genetic route the tumor had taken to escape cetuximab.
What this means for patients
For people whose colorectal cancer stops responding to cetuximab, this study offers cautious optimism. It suggests that even after tumors become resistant to EGFR-targeted therapy, many cancer cells remain primed to die if a key survival protein, BCL-xL, is blocked. While BCL-xL inhibitors can have side effects, especially on blood platelets, the work points toward combination and dose-optimizing strategies that could limit toxicity while exploiting a shared Achilles’ heel of hard-to-treat tumors. In future, drugs that disarm BCL-xL might form the backbone of new second-line treatments for cetuximab-refractory colorectal cancer, less dependent on the tumor’s shifting mutational landscape.
Citation: Asmanidou, S., Thiel, J., Ekstrom, T.L. et al. BCL-xL as a therapeutic target in cetuximab-refractory colorectal cancer. Cell Death Dis 17, 187 (2026). https://doi.org/10.1038/s41419-026-08434-5
Keywords: colorectal cancer, drug resistance, cetuximab, BCL-xL inhibition, apoptosis