Clear Sky Science · en
Mitochondrial DNA drives NLRP3-IL-1β axis activation in microglia by binding to NLRP3, leading to neurodegeneration in Parkinson’s disease models
Why this matters for Parkinson’s disease
Parkinson’s disease is best known for tremors and slowed movement, but beneath these symptoms lies a complex battle inside the brain. This study uncovers how tiny fragments of genetic material from damaged mitochondria in brain immune cells can ignite inflammation that gradually kills the dopamine-producing neurons needed for smooth movement. Understanding this chain reaction opens up new, very specific targets for drugs that might slow or prevent Parkinson’s-like damage.
Brain immune cells and powerhouses under stress
The brain contains not only neurons but also microglia, its resident immune cells. In Parkinson’s disease, these microglia often become overactive, releasing toxic molecules and inflammatory signals that harm nearby neurons. The authors focused on mitochondria, the “power plants” of cells, which carry their own DNA. When mitochondria are injured—by toxins, aging, or other stresses—their DNA can become oxidized, a kind of chemical damage caused by reactive oxygen species. Because mitochondrial DNA resembles bacterial DNA, these damaged fragments can act like alarm signals inside the brain, alerting the immune system and potentially driving chronic inflammation.
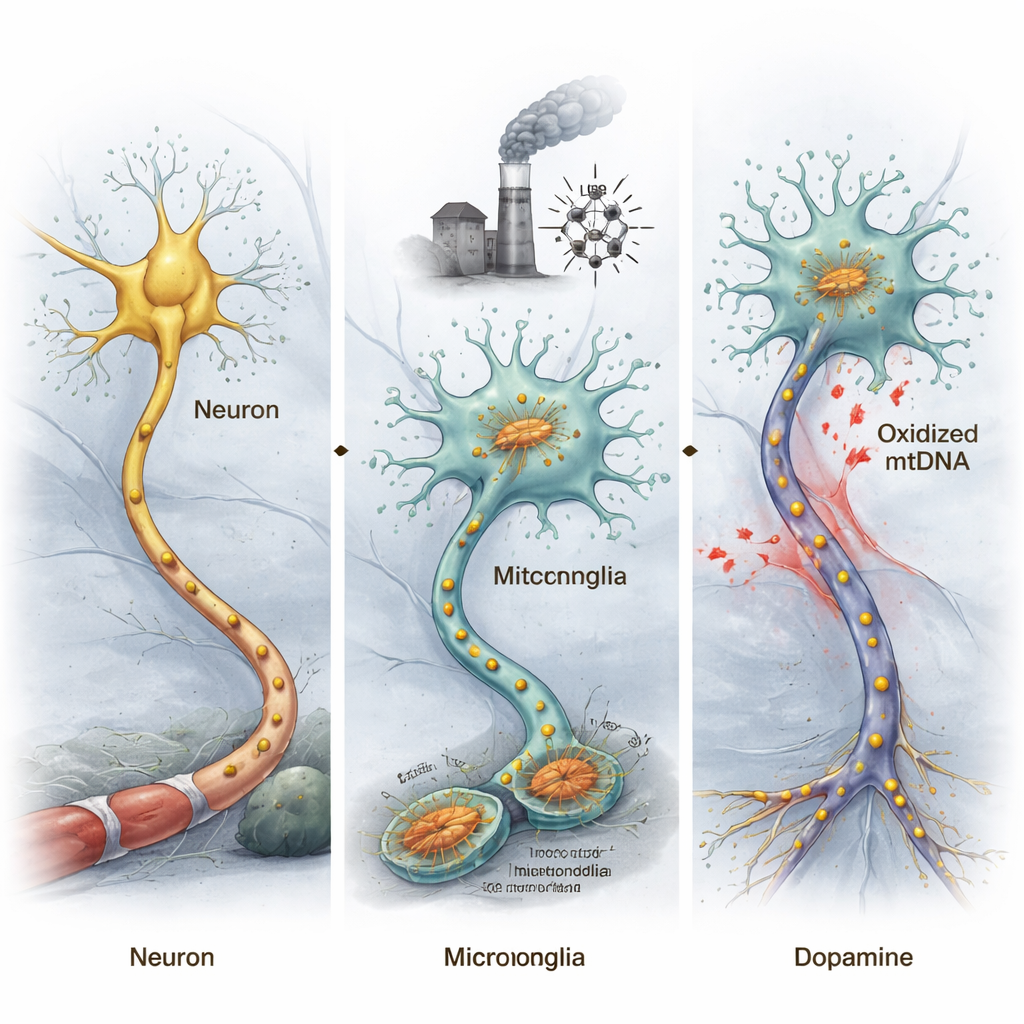
Damaged mitochondrial DNA can push mice toward Parkinson’s-like disease
To test whether oxidized mitochondrial DNA (ox-mtDNA) can actually trigger Parkinson’s-like changes, the researchers extracted ox-mtDNA from stressed microglial cells and injected it directly into a brain region that normally contains dopamine-producing neurons important for movement. Mice exposed to this ox-mtDNA showed decreased movement in behavioral tests and a loss of these dopamine neurons, echoing core features of Parkinson’s disease. By sorting out neurons and microglia from the midbrain and analyzing their gene activity, the team found that genes linked to Parkinson’s disease became more abnormal in neurons, while inflammation-related genes were strongly activated in microglia.
A molecular alarm switch inside microglia
The study zeroed in on a protein complex in microglia called NLRP3, part of a larger assembly known as the inflammasome. When switched on, NLRP3 helps activate caspase-1, an enzyme that cuts the inflammatory molecule IL-1β into its active, harmful form. Both in mice and in cultured cells, the combination of an inflammatory priming signal (LPS, a bacterial component) and the pesticide-like toxin rotenone caused mitochondria in microglia to release oxidized mtDNA into the cell fluid. This release coincided with stronger activation of NLRP3, more cleaved (active) caspase-1, and higher IL-1β levels. Medium taken from these overactivated microglia was directly toxic to dopamine-like neurons grown in a dish, lowering their survival and the level of their key marker, tyrosine hydroxylase.
Direct binding: how oxidized DNA flips the NLRP3 switch
Beyond simple correlation, the authors showed that ox-mtDNA is not just present during inflammation—it appears to bind physically to NLRP3 and turn it on. When they blocked mitochondrial DNA release with a drug, activation of caspase-1 and IL-1β dropped. Conversely, directly delivering oxidized mtDNA into microglia strongly boosted NLRP3 signaling, more than non-oxidized mtDNA. Using biochemical pull-down experiments, they detected mtDNA, rich in oxidative damage markers, attached to NLRP3. Computer modeling and protein chemistry revealed that ox-mtDNA binds a positively charged, flexible segment of NLRP3 (an “intrinsically disordered region” spanning amino acids 180–187). When this critical stretch was removed from NLRP3, its ability to bind ox-mtDNA and activate caspase-1 was greatly reduced.
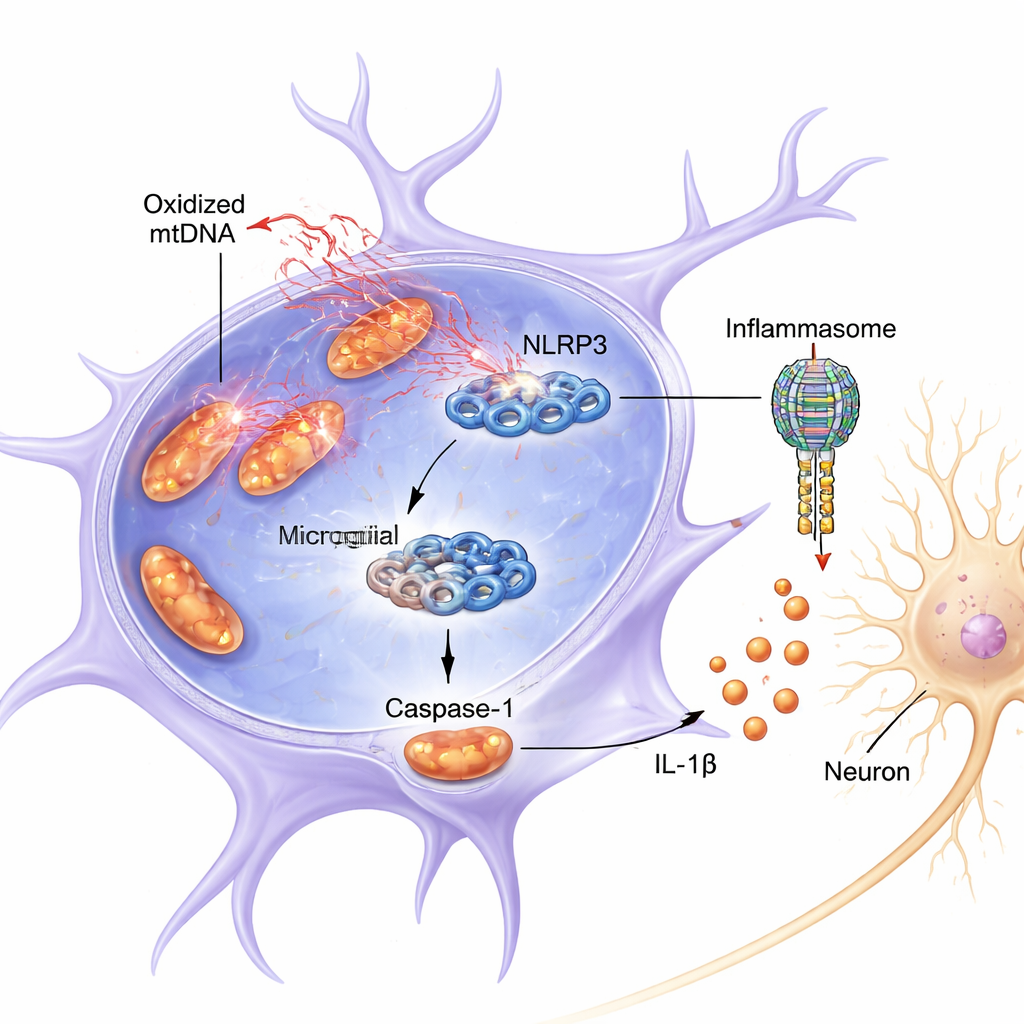
Turning off the inflammasome spares neurons
The researchers then asked whether interfering with NLRP3 could protect neurons. In cultured microglia, genetically reducing NLRP3 sharply lowered caspase-1 and IL-1β activation after LPS and rotenone treatment, and the conditioned medium from these cells was far less toxic to dopamine-like neurons. In mice, a selective NLRP3 inhibitor (MCC950) reduced microglial activation, preserved dopamine neurons, and lowered inflammasome-related proteins in models driven either by LPS plus rotenone or by injected mtDNA. Together, these experiments show that the ox-mtDNA–NLRP3–IL-1β chain is not just associated with damage; it is required for much of the observed neuron loss.
What this means for future Parkinson’s therapies
For non-specialists, the key message is that damaged mitochondrial DNA leaking from brain immune cells can directly latch onto an internal “alarm switch” (NLRP3), causing a wave of inflammation that injures dopamine neurons and drives Parkinson’s-like disease in models. Because this interaction depends on a short, defined segment of the NLRP3 protein and on the oxidized nature of the mitochondrial DNA, it offers precise new drug targets. Therapies that prevent mitochondrial DNA damage or escape, block its binding to NLRP3, or inhibit NLRP3 itself could, in principle, cool this inflammatory cascade and slow the progression of Parkinson’s disease.
Citation: Gan, Q., Fu, X., Zhou, T. et al. Mitochondrial DNA drives NLRP3-IL-1β axis activation in microglia by binding to NLRP3, leading to neurodegeneration in Parkinson’s disease models. Cell Death Dis 17, 213 (2026). https://doi.org/10.1038/s41419-026-08424-7
Keywords: Parkinson’s disease, microglia, mitochondrial DNA, NLRP3 inflammasome, neuroinflammation