Clear Sky Science · en
Aberrant maintenance of developmental transcription factor PAX6 promotes neuronal cell death via JNK3 signaling
Why this research matters for vision
Glaucoma is a leading cause of permanent blindness, largely because the nerve cells that carry visual information from the eye to the brain slowly die. Many treatments lower eye pressure, but people can still lose sight even when pressure is well controlled. This study asks a deeper question: what makes these retinal nerve cells decide to die when they are under stress, and can we switch off that decision at the level of gene control inside the cell nucleus?
A stressed retina under attack
At the heart of glaucoma and related eye diseases is the slow loss of retinal ganglion cells (RGCs), the output neurons of the eye. These cells are vulnerable to many kinds of stress, including toxic levels of the brain messenger glutamate, which overactivates NMDA receptors and triggers damaging calcium overload. The researchers used a well-established mouse model in which a small amount of NMDA is injected into the eye, selectively injuring RGCs while leaving other retinal layers largely intact. They confirmed that this treatment did not change eye pressure, but did cause typical signs of programmed cell death in RGCs, such as release of cytochrome c from mitochondria and appearance of TUNEL-positive nuclei.
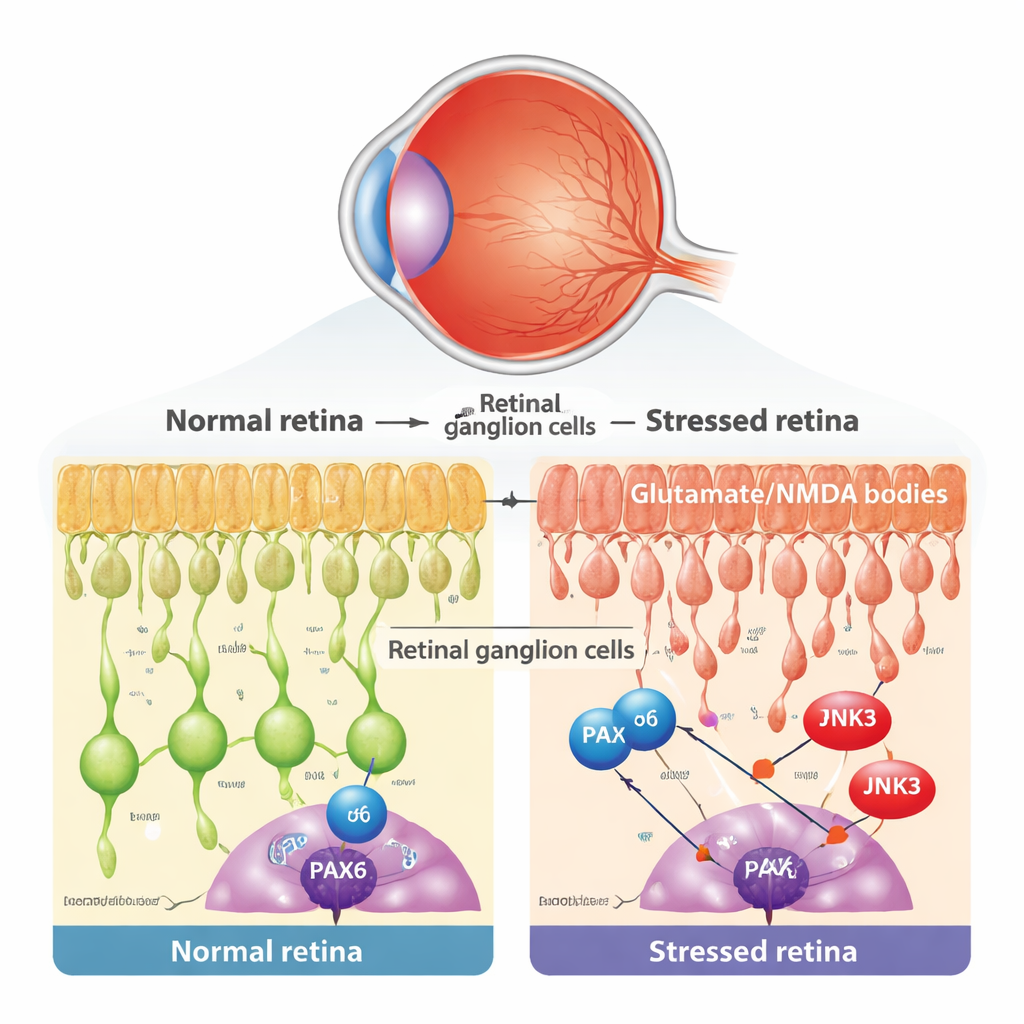
A development gene that refuses to retire
During early development, a gene regulator called PAX6 acts as a master architect of the eye, guiding how different retinal cells are born and wired. Conventional wisdom says such developmental programs mostly shut down in adulthood. By reanalyzing single-cell RNA sequencing data from both mouse and human retinas, the team found that PAX6 is in fact strongly and selectively maintained in mature RGCs and certain interneurons. Using microscopic staining, they showed that in the layer where RGCs reside, PAX6 is largely present in ganglion cells rather than neighboring amacrine cells. This raised an intriguing possibility: in adult disease, an old developmental program might be co-opted and turned into a driver of degeneration.
From guardian to executioner: PAX6 switches roles
To test whether PAX6 helps RGCs survive or die under stress, the scientists used a gene therapy–like approach. They delivered a viral vector carrying a small RNA that specifically knocks down PAX6 in the retina, and then exposed the eyes to NMDA. Compared with control-treated eyes, PAX6-depleted retinas showed far fewer apoptotic RGCs and much less mitochondrial damage, indicating that PAX6 is required for full-blown cell death in this model. Genome-wide RNA sequencing revealed that many pro-death genes, particularly those involved in mitochondrial damage and caspase activation, were strongly induced by NMDA in normal mice but were blunted when PAX6 was silenced. In other words, PAX6 helps turn on a network of genes that push RGCs over the edge.
The stress kinase that flips the PAX6 switch
How does stress activate PAX6 without increasing its amount? The team focused on JNK3, a stress-responsive enzyme found mainly in neurons. Under NMDA injury, JNK3 moved into the nucleus of RGCs and physically associated with PAX6. Biochemical "test tube" experiments using purified proteins showed that JNK3 can directly add phosphate tags to PAX6, and this reaction was blocked by a JNK inhibitor. In mice lacking the Jnk3 gene, NMDA no longer produced the same pattern of PAX6 phosphorylation. Chromatin mapping (ChIP-seq) and targeted DNA-binding assays revealed that, under stress, phosphorylated PAX6, together with JNK3, becomes more strongly bound at the control regions of key pro-apoptotic genes such as Bax and Gadd45a, boosting their activity. When either PAX6 was knocked down or JNK3 was genetically removed, this binding and the corresponding pro-death gene activation were sharply reduced.
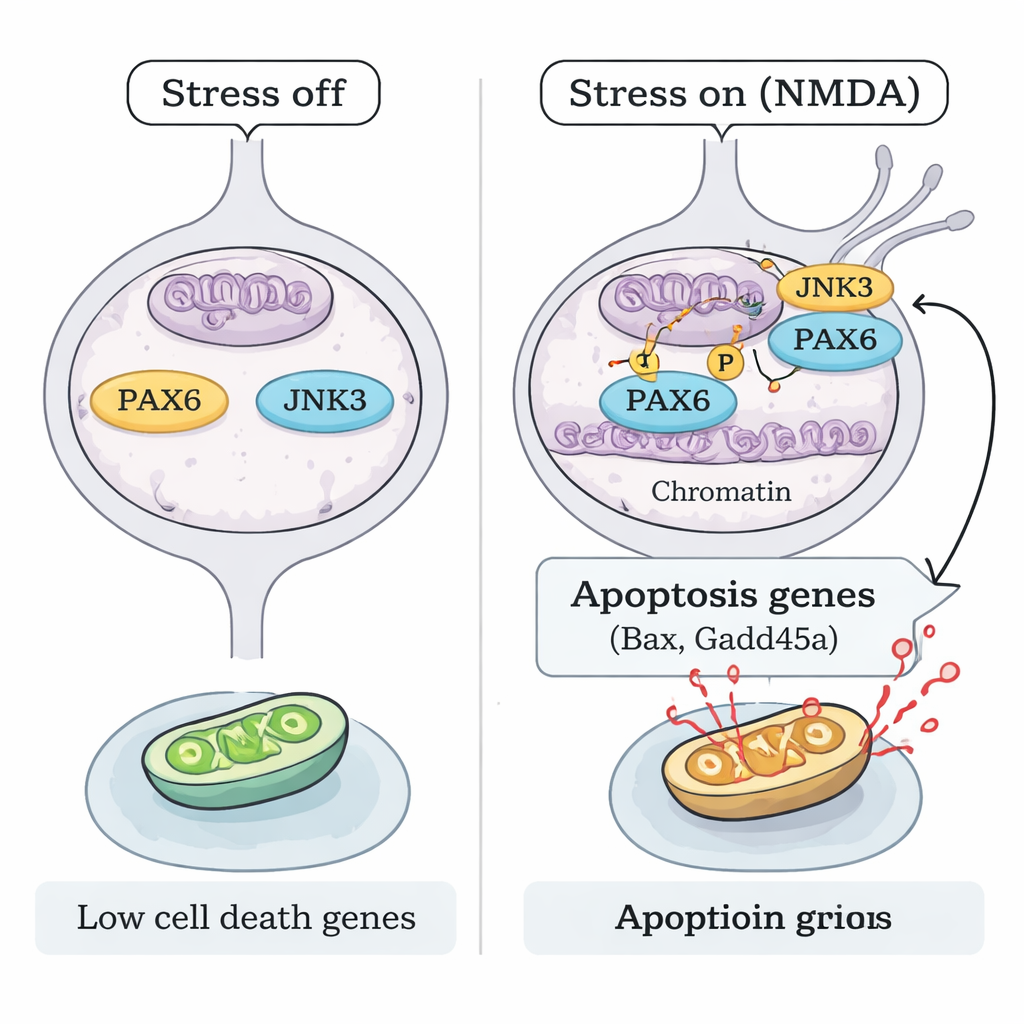
Turning off the death program to protect sight
Finally, the researchers asked whether blocking this JNK3–PAX6 axis is enough to protect vision-critical cells. In both PAX6 knockdown mice and JNK3-deficient mice, RGCs were significantly preserved after NMDA exposure, with fewer dying cells and healthier retinal structure. This points to a clear mechanistic model: under excitotoxic stress, JNK3 phosphorylates the persistently expressed PAX6, converting it from a developmental builder into a potent activator of a cell-death gene program in adult RGCs. Interrupting that link—by silencing PAX6 or disabling JNK3—keeps many of these neurons alive. For patients, this work suggests that future glaucoma therapies might go beyond lowering eye pressure and directly target the genetic switches that decide whether retinal neurons live or die.
Citation: Kim, JY., An, MJ., Kim, J. et al. Aberrant maintenance of developmental transcription factor PAX6 promotes neuronal cell death via JNK3 signaling. Cell Death Dis 17, 161 (2026). https://doi.org/10.1038/s41419-026-08417-6
Keywords: glaucoma, retinal ganglion cells, PAX6, JNK3, neurodegeneration