Clear Sky Science · en
Computational variant predictors for pharmacogenomics: from evaluation of single alleles to assessment of adverse drug reactions to antidepressants
Why your genes matter for antidepressant safety
When two people take the same antidepressant, one may feel better with few side effects while another struggles with serious problems, including drug toxicity. This study explores whether computer programs can read tiny differences in our DNA to predict who is likely to process antidepressants safely and who may be at higher risk for harmful reactions, potentially making everyday prescribing both safer and more precise.

From rigid labels to flexible genetic scores
Today, many clinics rely on a system called “star alleles,” which groups known DNA variants in drug-processing genes into a few broad function categories, such as normal or reduced activity. This approach has helped guide treatment, but it breaks down when a person carries rare or never-before-seen variants, or complex combinations of changes that are not on the official lists. The authors argue that this is a major blind spot: most pharmacogenetic variants are rare, and a sizable share of variation in how people handle medicines remains unexplained by current labels.
Testing smarter tools on known and new variants
The team evaluated ten computational tools that score how damaging a DNA change is likely to be, including two new frameworks they developed (PharmGScore and PharmMLScore). First, they asked whether these tools could reproduce the functional categories already assigned to 541 curated star alleles across eight key drug-processing genes. By adding up the scores of all variants within each haplotype, several tools matched or even exceeded the performance of the star system, with PharmGScore leading the pack. Next, they challenged the tools with data from high-throughput lab experiments on two important enzymes, CYP2C9 and CYP2C19, which process many medications. These experiments measured how thousands of individual variants affected enzyme activity and protein levels, most of which had never been seen in patients. Here again, the better tools, especially the pharmacogene-focused ensembles and CADD, accurately identified variants that severely impaired enzyme function.
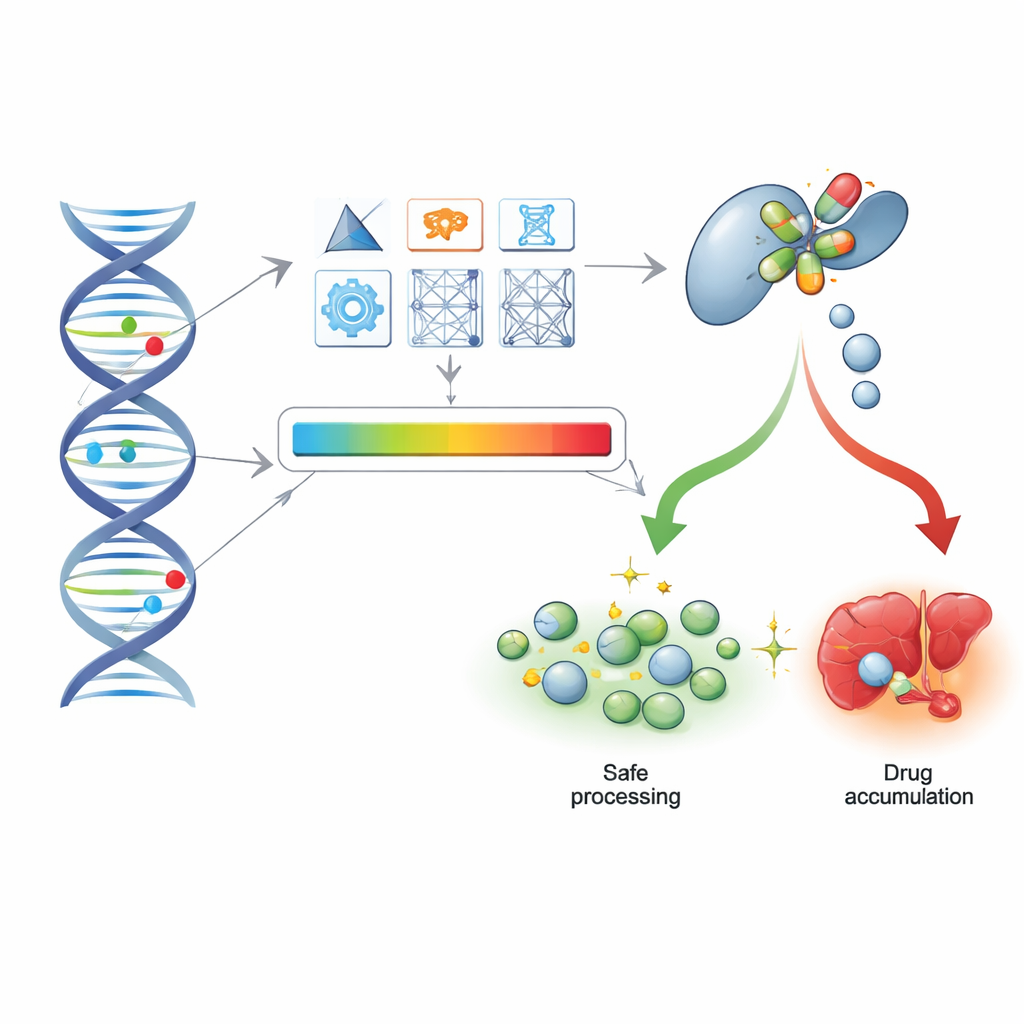
From DNA sequences to real-world patient records
To see if these computational scores hold up in everyday medicine, the researchers turned to exome sequencing data from more than 200,000 participants in the UK Biobank, along with their prescription histories and hospital records. They compared predictions from the tools against star allele calls for five major drug-processing genes and found that the best-scoring methods could largely recapture the same functional groupings, despite the fact that exome data miss some noncoding and structural changes. Importantly, the additive approach—summing up the impact of all variants in a gene—worked well enough to separate people with no-function genotypes from those with normal activity.
Spotting people at risk for serious antidepressant reactions
The authors then focused on antidepressant use and safety, zeroing in on the CYP2C19 enzyme, which helps break down several common depression medicines. Among more than 75,000 antidepressant users, they examined two outcomes: frequent switching of medications, as a rough marker of poor response, and hospital or death records indicating poisoning by antidepressants. While neither star alleles nor most scores showed a strong or clear signal for treatment switching, they did reveal a meaningful pattern for severe adverse reactions. Carriers of damaging CYP2C19 variants had about a 20–35% higher odds of serious antidepressant poisoning codes in their records, whether classified by star alleles or by top-performing computational tools such as PharmGScore, PharmMLScore, and CADD. This relationship remained similar even when analyses were restricted to cases without documented self-harm.
What this could mean for future prescriptions
Overall, the study shows that thoughtfully designed computational predictors can reach the same level of accuracy as the traditional star allele system, while overcoming its biggest weakness: the inability to handle new, rare, or complex genetic variants. By translating raw DNA sequences into continuous risk scores that work across the whole genome, these tools could eventually allow clinicians to see beyond a short list of known genotypes and better anticipate who is at higher risk of serious antidepressant side effects. Before they are used in routine care, more validation and integration with other clinical factors will be needed, but this work lays a strong foundation for safer, more personalized prescribing based on comprehensive genetic information.
Citation: Hajto, J., Piechota, M., Krätschmer, I. et al. Computational variant predictors for pharmacogenomics: from evaluation of single alleles to assessment of adverse drug reactions to antidepressants. Pharmacogenomics J 26, 8 (2026). https://doi.org/10.1038/s41397-026-00399-0
Keywords: pharmacogenomics, antidepressants, genetic variants, adverse drug reactions, computational prediction