Clear Sky Science · en
Visinin-like protein 1 disrupts calcium homeostasis and promotes atrial fibrillation in human and rodent models
Why this heart rhythm story matters
Atrial fibrillation is a common heart rhythm problem that raises the risk of stroke and heart failure. Many people live with it, yet doctors still struggle to prevent it or stop it from coming back after treatment. This study uncovers a previously overlooked protein in heart cells, called VILIP-1, that acts like a faulty calcium switch and helps drive atrial fibrillation in both people and animals. By pinpointing this switch and showing that existing drugs can dial it down, the work opens a fresh path toward safer, more targeted therapies.
A closer look at a chaotic heartbeat
In a healthy heart, electrical waves spread in an orderly fashion through the upper chambers, or atria, guiding each heartbeat. That order depends heavily on calcium, a charged mineral that surges in and out of heart cells to coordinate contraction and relaxation. In atrial fibrillation, this calcium traffic becomes unruly: extra leaks and swings in calcium levels spark stray electrical signals that can spiral into rapid, irregular rhythms. Doctors have known for years that calcium mishandling is central to this disorder, but the upstream triggers that start the chaos have remained murky.
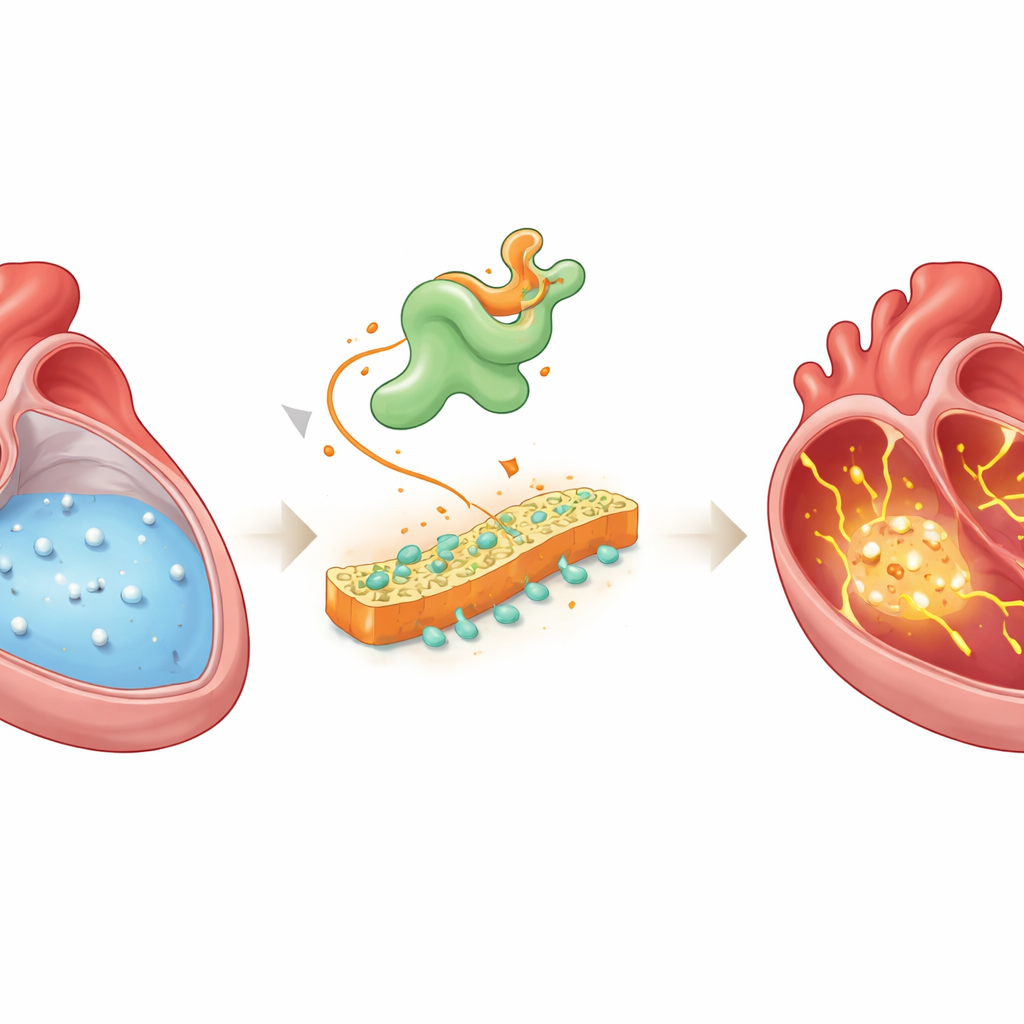
Finding a hidden troublemaker
The researchers combined several advanced approaches to search for molecules that link disturbed calcium handling to atrial fibrillation. They recorded calcium signals from hundreds of individual atrial cells in rats with pacing-induced atrial fibrillation, then sequenced the full set of genes active in those same cells. They also compared gene activity in atrial tissue from patients with atrial fibrillation to that of patients with normal rhythm. When they overlaid these datasets and tracked how cells changed along the path from healthy to diseased states, one gene repeatedly stood out: Vsnl1, which produces the protein VILIP-1, a calcium-sensing molecule previously studied mostly in the brain and pancreas, not in the heart.
How VILIP-1 disturbs calcium balance
In atrial tissue from both patients and animal models, VILIP-1 levels were significantly higher, and the protein was concentrated at the cell surface. To test whether this change was just a bystander or a driver, the team forced atrial heart cells in mice to produce extra VILIP-1. These mice did not show obvious structural damage to the heart, but their atria became much easier to push into atrial fibrillation during electrical pacing. Detailed electrical recordings showed more delayed afterdepolarizations—abnormal blips after each beat—and beat-to-beat alternations in action potential shape, classic signatures of unstable electrical behavior tied to calcium overload. High-resolution calcium imaging confirmed frequent spontaneous calcium waves and depleted internal calcium stores, indicating severe leakage inside the cells.
The critical partner on the cell surface
To understand how VILIP-1 causes this disruption, the scientists mapped which proteins interact with it inside heart cells. Using two complementary methods—pulling down binding partners from atrial tissue and labeling nearby proteins with a “biotin halo” in living cells—they narrowed in on NCX-1, the main sodium–calcium exchanger in the heart’s surface membrane. This exchanger normally helps eject calcium in exchange for sodium, but under certain conditions it can work in reverse and push calcium into the cell. The study showed that VILIP-1 physically binds NCX-1 and increases the number of NCX-1 molecules embedded in the membrane without raising total NCX-1 production. As a result, exchanger currents became larger, and blocking NCX-1 with a selective inhibitor reduced calcium waves and made atrial fibrillation harder to provoke in mice with excess VILIP-1.
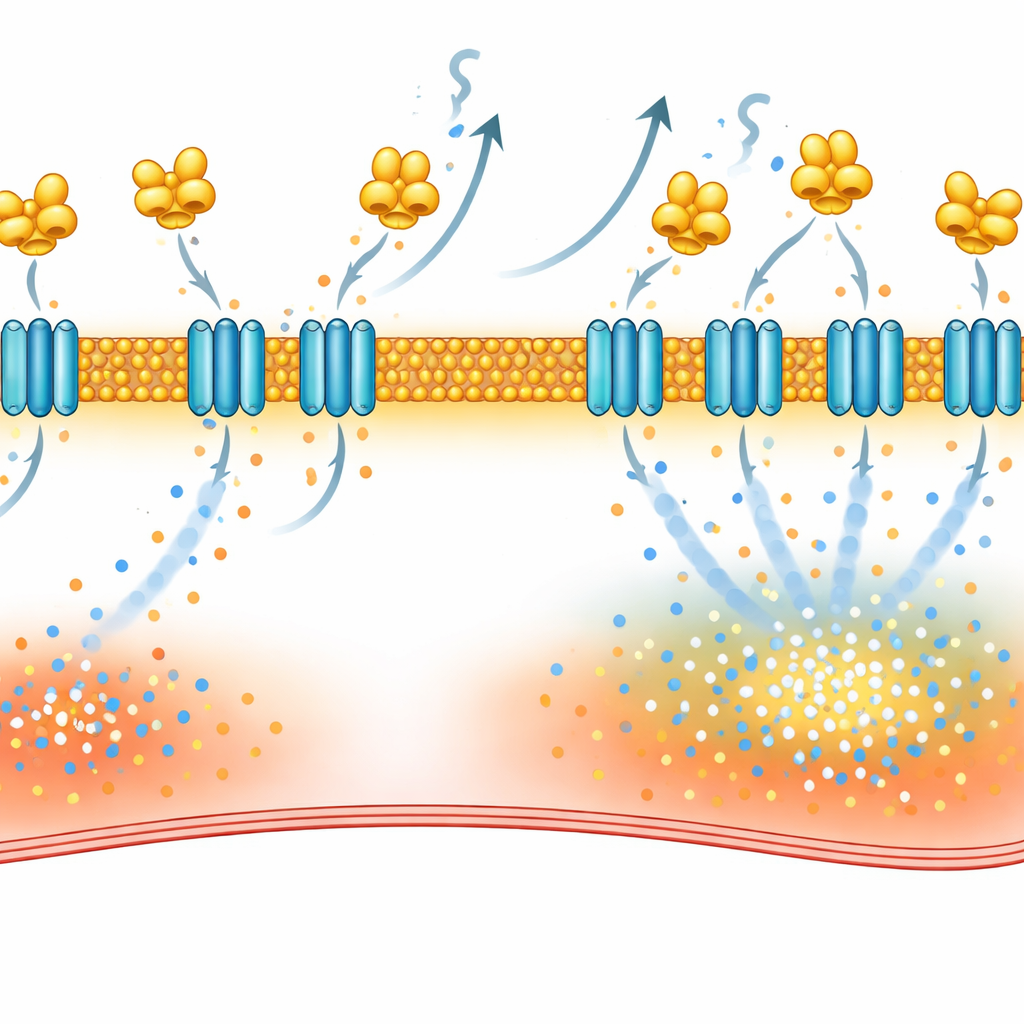
Turning off the faulty switch with existing drugs
VILIP-1 anchors to membranes through a fatty tag called myristate, which becomes exposed when calcium binds the protein. The team used desloratadine, previously shown to interfere with this tagging step, and found that it lowered NCX-1 at the cell surface, normalized exchanger currents, reduced calcium waves, and cut atrial fibrillation episodes in rat models. They also tested repaglinide, a diabetes drug known to bind related calcium sensors. Biophysical assays confirmed that repaglinide directly attaches to VILIP-1. In paced rats and in atrial tissue from patients with atrial fibrillation, repaglinide treatment reduced NCX-1 at the membrane, calmed calcium leakage in individual cells, and substantially lowered the ease with which atrial fibrillation could be induced.
What this means for people with irregular heartbeat
Altogether, the study outlines a self-reinforcing loop: rising calcium levels recruit VILIP-1 to the cell surface, where it boosts NCX-1, which in turn drives further calcium overload and sets the stage for atrial fibrillation. By interrupting this loop at the level of VILIP-1—either by blocking its fatty anchor or by binding its calcium-sensing core—existing drugs can restore a more stable calcium balance and reduce arrhythmia vulnerability in human and rodent heart tissue. While more work is needed to refine drug specificity and test these strategies in larger animal models and clinical trials, VILIP-1 now emerges as a promising new handle for preventing and treating this widespread and often stubborn heart rhythm disorder.
Citation: Xiong, K., Wang, G., Li, D. et al. Visinin-like protein 1 disrupts calcium homeostasis and promotes atrial fibrillation in human and rodent models. Sig Transduct Target Ther 11, 105 (2026). https://doi.org/10.1038/s41392-026-02615-6
Keywords: atrial fibrillation, calcium signaling, cardiac arrhythmia, sodium-calcium exchanger, therapeutic targets